
INTRODUCTION
Several major advances have improved our understanding of the molecular characteristics of hematological malignancies and led to the development of new therapeutic agents to eliminate these aggressive neoplasms. The discovery of the BCR-ABL1 fusion oncogene and defining the pathogenetic molecular mechanisms in chronic myelogenous leukemia (CML) have led to the development of BCR-ABL tyrosine kinase inhibitors such as imatinib, nilotinib, and dasatinib, which have improved the 10-year survival rate drastically in CML patients, from 20% to 85% [1-5]. In patients diagnosed with indolent or aggressive B-cell non-Hodgkin’s lymphoma, the use of the anti-CD20 antibody rituximab has resulted in improved survival [6]. These are only some of the most recognized examples of the breakthroughs that have occurred in the field of developing new therapies to treat hematological neoplasms. In spite of these discoveries, patients diagnosed with hematological malignancies continue to experience disease relapse and resistance to available treatment options, which suggests that the need to develop novel approaches that can be used alone or in combination with current therapeutic modalities to eradicate hematological neoplasms remains critical.
Numerous studies have concluded that the type I insulin-like growth factor receptor (IGF-IR) and its primary ligand IGF-I play significant roles in the establishment and progression of tumors, primarily by inhibiting apoptosis and inducing cellular transformation [7-10]. IGF-IR is also thought to aid malignant cells in acquiring anchorage-independent growth, giving the cells the ability to survive detachment and facilitate migratory processes for metastatic dissemination [11].
To date, there are several potentially effective IGF-IR inhibitors that have been tested in preclinical studies as well as in clinical trials enrolling patients harboring aggressive forms of solid cancers and hematological malignancies. Importantly, these IGF-IR inhibitors are well tolerated with minimal toxic effects in vivo [12]. The effects of IGF-IR have been studied to a great extent in solid tumors, including those of the breast, prostate, lung, ovary, skin, and soft tissues [13-17]. In contrast, less studies have been performed to thoroughly examine the function of IGF-IR in hematological neoplasms [18-24]. In this review, we discuss the current understanding of the role of IGF-IR signaling in cancer including hematological neoplasms. We also address the emergence of IGF-IR as a potential therapeutic target in the treatment of these aggressive diseases.
The IGF Signaling System
Overview
The IGF signaling system plays significant roles in both embryonic and postnatal development as well as having important functions in normal adult physiology. The IGF system includes four receptors: insulin receptor (IR), IGF-IR, IGF-IIR, and the hybrid receptors consisting of one-half IR and one-half IGF-IR (Figure 1). These receptors interact with three main ligands: insulin, IGF-I, and IGF-II. IR, IGF-IR, and IGF-IIR have the strongest binding affinity for their respective ligands, whereas the binding of insulin to IGF-IR and IGF-I to IR is at least 100-fold less [25]. IGF-I and IGF-II signaling is mediated through IGF-IR; but IGF-I has at least 3-fold higher binding affinity than does IGF-II [25]. The IGF system also includes regulatory proteins, known as IGF binding proteins (IGFBPs) that regulate IGF signaling. Although up to 10 proteins have been described in the literature as IGFBPs, only IGFBP-1 thorough IGFBP-6 are considered true IGFBPs based on their conserved protein structure and high binding affinity for IGF-I and IGF-II [26].
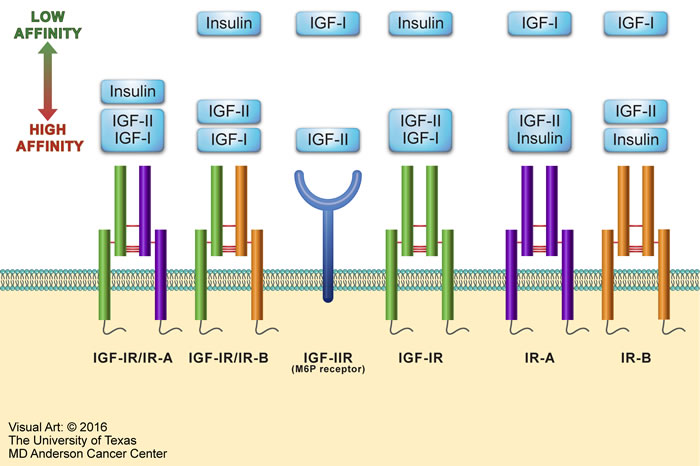
Figure 1: Overview of the IGF system. The IGF system consists of four receptors: IR, IGF-IR, IGF-IIR, and hybrid receptors. IR is expressed as two isoforms - IR-A and IR-B. IR-A has oncogenic potential, expressed predominantly in fetal tissues, and its expression declines during adulthood. IR-B is the physiologically expressed isoform in adult tissues. The IR-A or IR-B receptor makes one half of the hybrid receptors along with one half of the IGF-IR. The IGF system receptors interact mainly with three ligands: insulin, IGF-I, and IGF-II. Excluding IGF-IIR, these receptors possess tyrosine kinase activity. At the other hand, IGF-IIR (also known as mannose-6-phosphate [M6P] receptor) binds and removes circulating IGF-II to keep its free form at very low levels. The figure depicts IGF system ligands in order of their binding affinities to the different receptors. Ligands shown within the same rectangle have almost similar affinities to bind with a specific receptor. Ligands shown in separate yet close rectangles have slightly different receptor binding affinities. When the rectangles are widely separated, the ligands binding affinities are remarkably different.
IGF-IR
IGF-IR is a receptor tyrosine kinase that is structurally composed of two identical α subunits and two identical β subunits that are connected by disulfide bonds to form the functional homodimeric receptor complex (Figure 2). Each α subunit is entirely extracellular and contains a cysteine rich domain that forms the primary binding site for its ligands IGF-I, IGF-II, and, to a much lesser affinity, insulin. Each β subunit includes an extracellular domain, a 24-residue hydrophobic transmembrane domain, and a larger cytoplasmic region that shares 84% amino acid sequence identity with the IR [27]. Tyrosine 950 is the binding site for its substrates IRS-1 and SHC, among others. The intracellular region of the β subunit contains an ATP binding site at lysine 1003. It also contains a kinase domain encompassing the activation loop made up of three critical tyrosine residues at positions 1131, 1135, and 1136, which form part of the YXXXYY moiety (Y is a tyrosine and X is a non-tyrosine amino acid residues). It is important to note that the YXXXYY moiety is also present in other members of the IR family such as the anaplastic lymphoma kinase (ALK) and leukocyte tyrosine kinase receptors [28-30]. The tyrosine residues within the YXXXYY moiety become phosphorylated upon ligand binding-induced receptor dimerization. Residue 1136 in IGF-IR is particularly important in that it maintains the conformational stability of the β subunit.
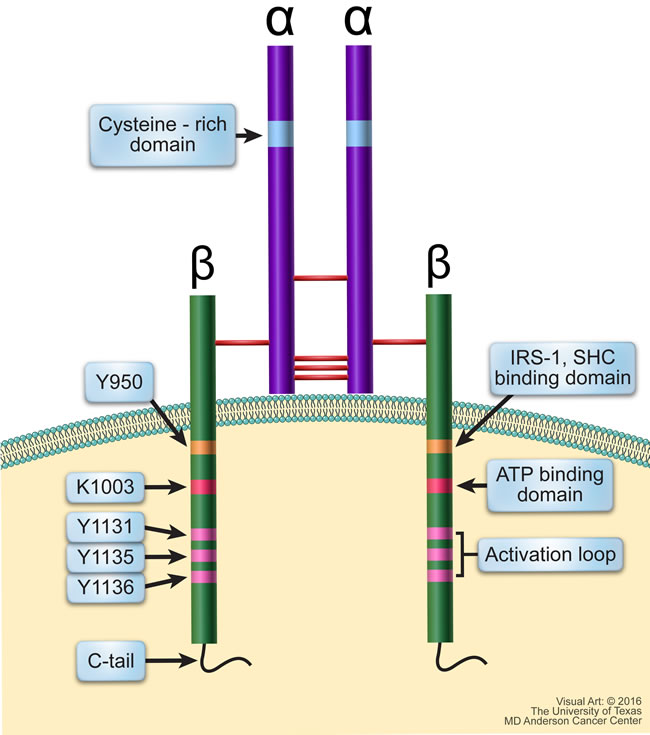
Figure 2: Schematic structure of IGF-IR. IGF-IR is a transmembranous homodimeric receptor tyrosine kinase that is composed of two identical extracellular α subunits and two identical transmembranous β subunits connected by disulfide bonds. The α subunit contains a cysteine-rich domain where the ligand binds. The cytoplasmic regions of the β subunits contain residues directly involved in IGF-IR signaling. Y950 is the binding site for its primary substrates including IRS-1 and SHC; Lys1003 is the ATP binding domain; and Tyr1131, Tyr1135, and Tyr1136 make up the activation loop of the kinase domain, which is followed by the C-terminus domain.
The C-terminus domain of IGF-IR contains several tyrosine and serine residues. These residues are phosphorylated, probably to induce mitogenic effects, but how their phosphorylation actually contributes to normal and malignant IGF-IR signaling is still not fully understood. The binding of IGF-IR to its ligands causes the phosphorylation of tyrosine residues located in the intracellular portion of its β subunit. Once phosphorylated, tyrosine 950 provides a docking site for IGF-IR substrates including IRS-1-4 and SHC proteins, which, in turn, act as docking sites. Upon substrate binding and phosphorylation of docking sites, downstream signaling is activated through the PI3K/AKT, MAPK, and STAT pathways and may stimulate cancer cell growth in an autocrine/paracrine manner [31-33] (Figure 3).
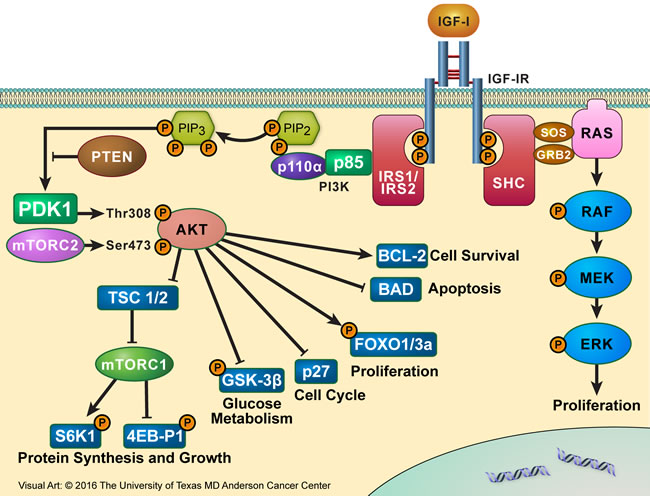
Figure 3: IGF-IR signaling partners. Binding of IGF-IR to its primary ligand, IGF-I, causes the phosphorylation of tyrosine residues present in the intracellular portion of its β subunit. Once phosphorylated, Y950 provides a docking site for its substrates IRS-1-4 and SHC proteins. Upon substrate binding, downstream signaling is activated through the IRS-1/PI3K/AKT/mTOR or SHC/RAS/MAPK pathway, leading to proliferative and anti-apoptotic effects, which culminate to oncogenesis and tumor cell survival.
Mouse models have revealed the importance of IGF-IR in prenatal and postnatal growth, especially in regards to genetic imprinting. The activation of IGF-IR during these stages improves the survival and increases the proliferation of mitosis-competent cells, resulting in the growth of tissues such as skin, bone, adipose tissue, and skeletal and cardiac muscles [34-38]. IGF-IR also plays roles related to the development of the mammary gland during the embryonic stage. In this regard, IGF-IR regulates the formation and proliferation of terminal end buds and contributes to ductal outgrowth and branching during puberty. In addition, it has been demonstrated that IGF-IR is necessary for normal epithelial proliferation and alveolar formation during pregnancy and lactation [39]. During embryonic development, the IGF-IR pathway is also involved in the development of limb buds. It has been previously shown that the abrogation of IGF-IR expression such as in Igf1r-null mice or the marked decrease in IGF-I levels leads to generalized organ hypoplasia, including developmental delays in bone ossification and abnormalities in the central nervous system [35, 38, 40-43]. Mice lacking the entire Igf1r gene typically exhibit a dramatic reduction in body mass, and they die at birth due to severe respiratory failure [40]. Mice carrying only one functional copy of Igf1r are born alive, but their body mass is ~45% lower than normal [40].
The aberrant activation of the IGF-IR pathway is also strongly associated with initiating cancer growth [12]. Within the past 20-30 years, evidence has emerged to support that IGF-IR overexpression plays a significant role in the development and progression of tumors, metastatic potential, and resistance to therapies. Studies have shown that IGF-IR induces its oncogenic effects through the inhibition of apoptosis and the induction of transformation and angiogenesis. Furthermore, IGF-IR regulates properties that cause malignant cells to acquire anchorage-independent survival, allowing them to endure detachment and migration, which is essential for metastatic dissemination [44].
Moreover, it has been documented that IGF-IR expression above a certain threshold can induce tumor cell progression that is independent of exogenous ligand stimulation. For example, when RM11A murine breast cancer cells that express high levels of exogenous IGF-IR were treated with IGF-I or IGF-II, the proliferative/survival potential of these cells was not enhanced, suggesting a ligand-independent mechanism [45]. At the other hand, wild-type RM11A cells expressing low levels of IGF-IR had a robust response to IGF stimulation, most likely because the activation of endogenously expressed low levels of IGF-IR is still dependent on IGF-I stimulation. Therefore, it was proposed that high expression of IGF-IR results in its constitutive activation independent of the presence of ligands [45].
It has also been shown that the level of IGF-IR expression, specifically a minimum number of its molecule, plays a key role in its transforming and oncogenic properties [46, 47]. For instance, in 3T3-like fibroblasts, the IGF-IR number needs to be greater than 20,000 receptors per cell to enable mitogenesis and transformation after stimulation with IGF-I [46]. Similarly, in a pancreatic cell line, PANC-1, the observed increase in the receptor number per cell from 40,000 to 100,000 is in the range required to enable IGF-I-stimulated growth and therefore may be of central importance for pancreatic tumor growth [47]. Based on these findings, it was concluded that a relatively small increase in the number of receptors per cell above a certain threshold could induce the transformation effects of IGF-IR.
Studies on IGF system in normal epithelial and cancerous cells such as those of the breast have found that IGF-IR is activated in one of two ways through IGF-I: (1) hepatic production of IGF-I can increase serum IGF-I, which in turn may act in an autocrine manner as a stimulatory molecule to induce the proliferation of normal as well as cancer cells; and/or (2) local production of IGF-I by stromal cells that surround the tumorigenic cells serves as a paracrine stimulator of the IGF-IR signaling [48, 49]. It has also been proposed that the interactions between IGF-I and IGF-IR enhance tumor cell survival through stimulation of other oncogenic signaling mechanisms. For instance, in prostate cancer cells, IGF-I is thought to indirectly stimulate the androgen receptor via crosstalk with IGF-IR to induce the transformation of prostatic epithelial cells [50, 51]. The oncogenic effects of IGF-I/IGF-IR interactions have also been suggested to exist in colon cancer. IGF-IR is highly expressed in colon cancer, compared to normal colonic mucosa, and IGFs in colonic malignant mucosa could exert their effects via IGF-IR in a paracrine manner [51-53].
In contrast to the widely studied solid tumors, not many studies have examined the role of IGF-IR in hematological neoplasms, and most of these studies focused on plasma cell myeloma [54, 55]. Other studies also addressed the role of IGF-IR signaling in acute myelogenous leukemia (AML) and acute lymphoblastic leukemia (ALL) [56-64]. In addition, studies from our lab and others’ have demonstrated the contributions of IGF-IR signaling to the pathogenesis of other hematological neoplasms, including T-cell lymphoma, mantle cell lymphoma, chronic lymphocytic leukemia (CLL), and CML [20-24, 65].
IGF-IIR
Also, there are studies showing the involvement of IGF-IIR in cancer development and progression. IGF-IIR, known as the cation-independent mannose-6-phosphate receptor, acts as a reservoir to clear IGF-II from the extracellular environment and cell surface through endocytosis and lysosomal degradation, thereby inhibiting IGF-II-induced tumor growth signaling [66-68]. Therefore, IGF-IIR is considered a tumor suppressor protein. It has been demonstrated that specific alterations in IGF-IIR lead to the progression of a transformed phenotype. For example, microsatellite instability of IGF-IIR has been documented in cancers of the prostate, breast, endometrium, and gastrointestinal tract [69-71]. In addition, loss of heterozygosity of IGF-IIR has been described in liver, lung, ovarian, prostate, head and neck, and breast cancers [72-76]. While loss of heterozygosity in tumor samples suggests the loss of IGF-IIR function and the enhancement of the progression toward a transformed phenotype, the detection of mutations in IGF-IIR that disrupt ligand binding at the cell surface is another mechanism by which IGF-IIR loses its function [77, 78].
IGF-I and IGF-II
IGF-I is a single-chain peptide that shares 62% and 40% amino acid sequence identities with IGF-II and proinsulin, respectively [79]. IGF-I is synthesized in a variety of tissues and cultured cell types, suggesting that this protein may have autocrine and paracrine effector functions, unlike insulin, which is stored within cells of a particular tissue (pancreas) and has mainly autocrine effects [38, 80]. Liver secretion of IGF-I is regulated by the growth hormone (GH), which signals peripheral tissues to grow, whereas insulin is tightly regulated by glucose uptake [80, 81]. Therefore, insulin is primarily associated with metabolic effects, whereas IGF-I is essentially a growth factor and an anabolic agent.
IGF-I is a ~7.6 kDa 70 amino acid single chain polypeptide that functions as the primary ligand for IGF-IR. There is increasing evidence that IGF-I might provide a major link between IGF-IR and the development of cancer through its regulatory effects on cell proliferation, differentiation, and apoptosis. Although cancer cells do not necessarily secrete IGF-I, high concentrations of circulating IGF-I, which is secreted by the liver, have been found to associate increased cancer risk in children and adults as well as to contribute to the growth, maintenance, and progression of cancer [82-86]. Mouse models have shown reduced proliferation of androgen-dependent prostate cancer cells in IGF-I-deficient hosts relative to control hosts [87]. In contrast, spontaneous neoplasia occurs in prostate epithelium from Igf1 transgenic mice [88]. In breast cancer, it has been shown that elevated IGF-I levels can reduce the synthesis of sex hormone-binding globulin, which can lead to an increase in the bioavailability of estrogen. Estrogen has been demonstrated to induce the expression of IGF-IR as well as IRS-1 and IRS-2, which synergistically potentiate the activation of MAPK [89].
IGF-II is a ~7.5 kDa 67 amino acid single chain mitogenic polypeptide that is produced mainly by the liver [68, 90-93]. It is also synthesized by other tissues, such as muscle and placenta, in which it elicits its effects through an autocrine or paracrine manner, similar to IGF-I [94, 95]. Physiologically, IGF-II is involved in the regulation of fetal development (it is highly expressed during embryonic stages), cell growth, differentiation, and metabolism [90, 96-104]. Unlike IGF-I that is tightly regulated by GH, genetic factors, such as loss of imprinting, play a significant role in the regulation of IGF-II, which causes an overabundance of IGF-II and subsequent augmentation of its signaling [105, 106]. Loss of imprinting has been demonstrated in a variety of cancers including breast, ovarian, and esophageal, and kidney cancers. Furthermore, IGF-II can also be regulated at the transcriptional level either through direct binding of transcription factors on consensus motifs or through hormone-induced transcription [107-112]. IGF-II is believed to induce its effects mainly through IGF-IR, IR isoform A (IR-A), and IGF-IR/IR-A hybrid receptors. Similar to IGF-I, the binding of IGF-II to IGF-IR induces downstream signaling via the PI3K/AKT and MAPK pathways [113]. IGF-II is cleared from the extracellular environment through binding with IGF-IIR. The overexpression of IGF-II in cancer cells has been demonstrated using in vitro and in vivo experimental approaches as well as in cancer patients’ specimens [114-120]. Moreover, in vivo studies showed that IGF-II overexpression results in more aggressive tumors, while animals with low expression of IGF-II or treated with IGF-II-neutralizing antibodies had lower tumor burdens and prolonged survival [114, 117, 118, 121].
Role of IGF-IR Signaling in Hematological Malignancies
Plasma cell myeloma
Plasma cell myeloma (PCM) accounts for approximately 10% of hematological malignancies with an incidence of four cases per 100,000 people a year [122]. The average age at diagnosis is 68 years, and the incidence increases with age advancement. The development of PCM occurs in steps. Normal plasma cells are usually arrested in the G1 phase of the cell cycle; however, malignant PCM cells exhibit uncontrolled cell cycle progression. PCM cells initially reside in the bone marrow, where they interact with stromal cells that express integrins and secrete cytokines to potentiate migration, adhesion, and proliferation of the PCM cells, and enhance surrounding angiogenesis [123-125]. The plasma cells then ultimately invade the basement membrane of the bone marrow and may undergo widespread dissemination. These processes are thought to be highly dependent on interleukin-6 (IL-6), as targeting IL-6/IL-6 receptor (IL-6R) signaling decreases PCM tumor growth in vitro and in vivo [126-129].
In addition to IL-6, it has also been shown that IGF-I induces the growth and development of PCM through activation of IGF-IR. For instance, some earlier studies demonstrated that IGF-I is capable of increasing DNA synthesis as well as enhancing the viability and proliferation of several PCM cell lines. The effects of IGF-I were reversed when an anti-IGF-IR blocking antibody was additionally used [130, 131]. In further support of an important role of IGF-I/IGF-IR signaling in PCM, studies from different labs showed that IGF-IR and IGF-I are highly expressed in PCM, and that the IGF-I/IGF-IR signaling axis, at least partially through an autocrine cascade, contributes significantly to the survival and progression of PCM [54, 132]. Different models have been proposed to explain the functional relationship between IGF-I/IGF-IR and IL-6 in PCM. In one model, the effects of IGF-I and IL-6 on PCM cells appeared to be executed independently. Indeed, anti-IGF-IR/IGF-I antibodies failed to block the stimulatory effects of IL-6. In a similar fashion, anti-IL-6/gp130 antibodies were not capable of reversing the effects of IGF-I [133]. Importantly, IGF-I and IL-6 were found to induce “additive” effects on PCM cell survival and proliferation. In support of this idea, IGF-I/IGF-IR axis induced its effects predominantly through the phosphorylation of IRS-1 protein, which is considered a major substrate of IGF-I/IGF-IR signaling [31, 134, 135]. Moreover, IL-6/gp130 promoted the survival of PCM cells through the phosphorylation of STAT3. Both signaling pathways, however, efficiently activated the RAS/ERK/MAPK survival-promoting system [133]. In another model, IL-6 was found to phosphorylate/activate IGF-IR receptor in PCM cell lines [136]. Interestingly, IL-6 stimulation of PCM cells was associated with co-localization of the IL-6Rα with IGF-IRβ at lipid rafts.
IGF-I/IGF-IR axis supports the survival of PCM cells not only via signaling through IRS-1 but also by singling through IRS-2. The IRS-2/PI3K/p70S6K pathway was found to be either constitutively activated or instead activated via upstream interactions with IGF-I/IGF-IR [19]. When IRS-2/PI3K/p70S6K pathway is constitutively activated, the direct binding of IRS-2 with IGF-IR, regardless of IGF-I stimulation, induces downstream effects that lead to tumor cell proliferation and inhibition of apoptosis [19].
IGF-IR signaling inhibits apoptosis and induces proliferation of PCM cells
In PCM, IGF-I/IGF-IR signaling inhibits apoptosis and induces tumor cell proliferation through the activation of two distinct pathways - PI3K/AKT and MAPK. Which of the two processes - apoptosis or cell proliferation - is promoted appears to depend on which pathway is activated. For instance, IGF-IR suppresses apoptosis through inhibition of the release of cytochrome C from the mitochondria after the activation of the PI3K/AKT pathway. Activated AKT phosphorylates the pro-apoptotic protein BAD. Upon phosphorylation, BAD becomes dissociated from anti-apoptotic BCL-2 and sequestered by the adaptor protein 14-3-3. These processes prevent apoptosis from occurring in PCM cells [137, 138].
One possible mechanism for the activation of the PI3K/AKT pathway in PCM is the lack of the PTEN tumor suppressor protein, a negative regulator of PI3K/AKT activity [139, 140]. The anti-apoptotic effect of PI3K/AKT signaling after IGF-I stimulation was found to be reversed upon overexpression of PTEN in PCM cell lines lacking PTEN, suggesting that the loss of PTEN is also responsible for uncontrolled IGF-IR-induced AKT activity in these cells [139, 140].
Whereas the activation of the PI3K/AKT pathway by IGF-IR appears to be involved primarily with inhibition of apoptosis in PCM, activation of the MAPK pathway is believed to be principally involved with inducing cell proliferation [138]. Ligand binding of IGF-I to IGF-IR allows the phosphorylation of the SHC substrate, which in turn leads to activation of RAS that subsequently activates the RAF kinase on its serine/threonine residues. As a result, activated RAF phosphorylates and activates MEK, which then phosphorylates and activates MAPK, leading to the proliferation of PCM cells [141].
In addition to the idea that distinct outcomes result from IGF-IR-induced activation of PI3K/AKT versus IGF-IR-induced activation of MAPK, more recent studies have revealed that “crosstalk” exists between PI3K/AKT and MAPK and that this crosstalk is critical for PCM tumor progression [141, 142]. In this model, binding of IGF-I to IGF-IR induces phosphorylation of RAF, MEK, and MAPK. However, inhibition of PI3K using the LY294002 inhibitor resulted in the inhibition of MEK and MAPK without interfering with the activation of upstream RAF [141]. This suggests that the PI3K pathway is capable of regulating the activation of MAPK, bypassing upstream activators of this pathway. Alternatively, using the MAPK inhibitor PD98059 did not have significant effects on the PI3K pathway, alluding to the idea that this crosstalk is mediated by PI3K, and not vice versa.
In a similar fashion, it was shown that rapamycin-induced mTOR inhibition decreased the serine phosphorylation of IRS-1, which was associated with a compensatory IGF-I downstream signaling via the PI3K/AKT pathway [142]. It was noted that this activation is highly dependent on IGF-IR signaling, because treating the PCM cells with an anti-IGF-IR antibody hindered the ability of rapamycin to activate AKT. Related observations were also shown in a different study utilizing the IGF-IR tyrosine kinase inhibitor picropodophyllin (PPP), where it was found that PPP significantly sensitized RPMI 8226 PCM cells to rapamycin and SB203580, a MAPK inhibitor [143]. These observations suggest a relationship and possible crosstalk between the mTOR and IGF pathways in PCM [142, 144].
In addition to the effects mediated by IGF-I, in vitro experiments have shown that IGF-II increases cell proliferation and protects dexamethasone-treated PCM cells from apoptosis [145]. Although this study did not examine in detail the interactions between IGF-II and IGF-IR, it was implied that at least the IGF-II-mediated proliferation of PCM cells was executed through IGF-IR. Whether the anti-apoptotic effects of IGF-II were also mediated through IGF-IR remains to be elucidated. Notably, IGF-II was almost as effective as IGF-I in protecting the PCM cells from dexamethasone-induced apoptosis [145].
IGF-IR induces adhesion, invasion, and migration of PCM cells
IGF-I/IGF-IR signaling is also involved in critical aspects of PCM dissemination, including adhesion, invasion, and migration. One of the principal features of malignant PCM cells during homing is their ability to transmigrate and adhere to the bone marrow microenvironment. In this regard, IGF-I acts as a chemo-attractant for PCM cells that increases their adhesion to the extracellular matrix glycoprotein fibronectin [146]. This effect is also dependent on the activation of IGF-IR downstream targets including PI3K/AKT [146]. In addition, IGF-I can rapidly and transiently induce the association between IGF-IR and β1 integrin, a heterodimeric membrane protein that mediates cell adhesion to the extracellular matrix, which also leads to further adhesion of PCM cells to fibronectin [146]. The phosphorylation of IGF-IR, IRS-1, and PI3K leads to the co-localization of IGF-IR and β1 integrin on lipid rafts in the cell membrane [146, 147].
It has also been shown that IGF-I triggers the polymerization of F-actin, induces phosphorylation of FAK and paxillin, and enhances β1 integrin’s association with these focal adhesion proteins, which enhances PCM homing to the bone marrow microenvironment [146, 148]. IGF-I induces the migration of PCM cells in an AKT-dependent or -independent manner, such as through the PKD or RhoA/PKC pathway [146, 149, 150]. This was further demonstrated when an anti-IGF-I antibody, anti-β1 integrin antibody or PI3K inhibitor abrogated IGF-I-induced transmigration [146].
Acute and chronic leukemia and myelodysplastic syndromes
Upregulation of IGF-I/IGF-IR signaling is prevalent in several types of leukemia, including AML, B-ALL, T-ALL, CML, and CLL.
Overexpression of IGF-IR has been reported in human AML cells and autocrine IGF-I production has been suggested to play a role in drug resistance in AML [18, 56, 57]. Furthermore, constitutive activation of the PI3K/AKT signaling pathway is believed to play crucial roles in the survival of AML cells [151, 152]. Constitutive activation of PI3K/AKT, at least in part, results from autocrine IGF-I release and activation of IGF-IR, which was shown in 70% of AML samples [18, 57]. Several in vitro studies using different IGF-IR inhibitors illustrated the therapeutic potential of targeting IGF-IR in AML. For example, NVP-ADW742, a small molecule inhibitor of IGF-IR, induces AKT dephosphorylation, which subsequently decreases p38 phosphorylation and downregulates antiapoptotic proteins such as BCL-2 in AML cells [60]. Inhibition of constitutive IGF-IR phosphorylation as well as downstream signaling through MEK and AKT was also demonstrated after treatment with BMS-536924 or BMS-554417, dual IGF-IR/IR inhibitors [59, 153]. In another study, the IGF-IR small molecule inhibitor NVP-AEW541 sensitized primary AML blasts and cell lines to etoposide-induced apoptosis through caspase-3 cleavage, in addition to inducing antiproliferative effects [56].
In addition, in AML, it has also been shown that mTOR inhibition using rapamycin overactivates PI3K/AKT by upregulating IGF-I/IGF-IR signaling, in a fashion similar to that seen in PCM. These effects appear to occur through enhancing not only IRS-1 expression but also IRS-2 expression. Using 19 AML samples with constitutive PI3K/AKT activation, the rapamycin-derivative inhibitor everolimus increased AKT phosphorylation by upregulating the expression of IRS-2 via an IGF-I/IGF-IR autocrine loop [58]. These findings suggest crosstalk between IGF-IR, PI3K/AKT, and mTOR in AML.
The myelodysplastic syndromes (MDS) arise from clonal hematopoietic stem cells that are characterized by atypical morphology, aberrant maturation, and specific molecular events. Approximately, one third of MDS patients experience progression to AML [154]. Decreased apoptosis of the hematopoietic cells plays an important role in the pathogenesis of MDS and probably in its transformation to AML [155-157]. Recently, IGF-IR has been shown to be involved in MDS pathogenesis and its transformation to AML. Compared with normal myeloid cells, IGF-IR protein and mRNA expression is enhanced in MDS cells, and this enhancement increases with progression to AML. In addition, in both MDS and AML, apoptotic signals are present only in IGF-IR-negative cells, suggesting that IGF-IR protects these cells from apoptosis [158].
Our group has discovered that IGF-IR also plays a role in CML [21]. In our study, IGF-IR was upregulated in CML cell lines, and the expression of IGF-IR appeared to be directly correlated with the progression of CML. For instance, IGF-IR was expressed in 30% and 25% of patients with chronic phase and accelerated phase CML, respectively, but the expression rate increased to 73% in patients with blast phase CML. Treatment of CML cells with PPP, an IGF-IR small molecule inhibitor, resulted in decreased cell viability and proliferation. In addition, inhibition of IGF-IR decreased activated/phosphorylated AKT and STAT5 as well as downregulated BCL-2, BCL-XL, and basal caspase-3 and was associated with apoptotic cell death. Inhibition of IGF-IR also resulted in cell cycle arrest at the G2/M-phase, which was mediated through upregulation of cyclin B1 and downregulation of cyclin E and pCdc2 [21].
In another recent study, the involvement of c-myb with IGF system in inducing the proliferation of CML cells was demonstrated [159]. C-myb plays an important role in the regulation of cell growth and differentiation and is highly expressed in malignant hematopoietic cells. Induced overexpression of c-myb causes a significant increase in IGF-I, IGF-II, and IGF-IR expression, and a decrease in IGFBP-3 expression, which promotes CML cell proliferation [159]. Of note is that anti-IGF-IR antibodies inhibited the c-myb-induced cell growth. The growth-promoting effects of c-myb were mediated through two major intracellular signaling pathways, AKT and ERK. The activation of AKT and ERK by c-myb was abrogated by IGF-IR and IGFBP-3 antibodies. These findings suggest that c-myb stimulates cell growth, in part, by regulating the expression of the components of the IGF system in CML.
The contribution of IGF-I/IGF-IR signaling to CLL has also been investigated. It was shown that IGF-I and IGF-IR participate in autocrine/paracrine loops to promote the survival of CLL cells [160]. High levels of IGF-IR protein and mRNA expression were detected in CLL cells and positively correlated with the expression of the antiapoptotic protein BCL-2. It was also shown that serum IGF-I was elevated in CLL patients, but the level of GH remained unchanged, suggesting that autocrine production of IGF-I by CLL cells results in increased levels of serum IGF-I, independently of GH, and can stimulate IGF-IR to promote CLL cell survival. Moreover, a recent study showed that the high expression of IGF-IR gene in CLL was associted with bad prognostic indicators including IGHV gene unmutated status, high CD38 expression, trisomy 12, and del(11)(q23) [161]. Furthermore, the increae in IGF-IR gene expression charectierized a subgroup of CLL patients with NOTCH1 mutation. In a separate study, 3 different IGF-IR inhibitors reduced CLL cell viability and induced apoptosis, even in the presence of the surrounding stromal cells, which provide a protective barrier [24]. Furthermore, it was found that sorafenib, a multikinase inhibitor, was able to decrease IGF-IR expression as well as decrease tyrosine kinase activity, suggesting IGF-IR is a target for sorafenib in CLL.
Significant work has also been accompalished in T-ALL and B-ALL to study the effects of not only IGF-IR but also IGFs. For instance, one study reported the characteristics of insulin, IR, IGF-I, IGF-II, and GH binding in 18 established T-ALL and B-ALL cell lines [162]. IGF-IR and IR were present in these cell lines; IGF-IR was preferentially expressed in T-ALL cell lines and IR was preferentially expressed in B-ALL cell lines. In addition, using binding assays, it was determined that there were different binding affinities for IGF-I, IGF-II, and insulin in the different cell lines. This was the first report to document the expression of these proteins in ALL. Another in vitro study used FL5.12, a murine pro B-ALL cell line, to further elucidate the main mechanism of IGF-IR signaling in this disease [163]. This cell line contains a Y1250F/Y1251F double-mutant IGF-IR, which causes loss of apoptotic induction in response to IGF-I withdrawal and deficient colony-forming capabilities and metastatic potential; however, these cells still display mitotic activity [163-166]. This study showed that the FL5.12 cells had impaired phosphorylation of JNK, SHC, and MAPK upon IGF-I stimulation, but IGF-I-induced phosphorylation of AKT was comparable to that in wild-type cells. Thus, this mutation hinders IGF-IR signaling by specifically abrogating the JNK, SHC, and MAPK pathways [163].
Some studies have aimed at identifying potential causes of IGF-IR overexpression in ALL. At least two groups have identified deregulated microRNAs that contribute to IGF-IR overexpression in T-ALL. It was shown that Notch1 repressed the expression of microRNA-223, which, in turn, was able to decrease the expression IGF-IR protein. However, increased expression of microRNA-233 alone did not significantly hinder cell growth, alluding to the possibility that, in addition to the decrease in IGF-IR expression, modifications of other survival molecules are probably required to fully suppress T-ALL [167]. Another study showed that the forced expression of microRNA-99a and microRNA-100 inhibited the expression of IGF-IR and mTOR, as well as the downstream oncogenic proteins MCL1 and the FK506-binding protein 51 in T-ALL cell lines. Importantly, microRNA-99a and microRNA-100 were significantly decreased in 111 ALL patients, and their decrease correlated with poor survival [168].
Evidence based on in vitro studies showed that targeting IGF-IR is a potential therapeutic strategy in ALL. For instance, one study showed that PPP efficiently decreased cell viability and induced G2/M-phase cell cycle arrest in Jurkat and Molt-3 T-ALL cell lines, with no effect on normal T lymphocytes. PPP was also able to increase the expression of cleaved caspase 3 and PARP and downregulate the anti-apoptotic proteins BCL-2 and BCL-XL, signifying apoptotic cell death. Furthermore, PPP decreased the phosphorylation of AKT and MAPK in T-ALL cells [64].
Myeloproliferative neoplasms (MPN) other than CML
This category encompasses a relatively uncommon group of hematological neoplasms that have specific clinicopathological and molecular characteristics. Polycythemia vera (PV), primary myelofibrosis, and essential thrombocythemia are the most frequently encountered MPN.
The role of IGF/IGF-IR signaling in MPN has been investigated in a limited number of studies, and most of these studies focused on PV. An earlier study showed that the early circulating erythroid progenitors collected from PV patients and maintained in serum-free medium possess remarkably enhanced sensitivity to IGF-I, much more pronounced than the sensitivity of control progenitor cells from healthy subjects, attesting to probable contribution of IGF-I/IGF-IR signaling to PV pathogenesis. Notably, the sensitivity of the cells from PV patients to IGF-I substantially exceeded their sensitivity to erythropoietin [169]. To investigate the molecular basis of IGF-I hypersensitivity, the same group found that basal and IGF-I-stimulated levels of IGF-IRβ subunit’s tyrosine phosphorylation were more enhanced in circulating mononuclear cells from patients with PV than in cells from normal individuals [170]. Furthermore, IGFBP-1 was shown to be significantly elevated in PV patients. Functionally, IGFBP-1 from these patients had pronounced stimulatory effects on erythroid burst formation in vitro [171]. Subsequent contradictory data showed no differences in IGF-IR phosphorylation levels in PV patients versus healthy controls [172]. However, this latter study further supported alterations in IGFBP levels including increased IGFBP-1, -2, and -4, and decreased IGFBP-3 in these patients. More recently, the expression of JAK2V617F mutant, a hallmark molecular event in PV, was found to render Ba/F3 cells hypersensitive to IGF-I stimulation, providing a molecular evidence that connects IGF-I/IGF-IR signaling to PV [173].
Arsenite-inducible RNA-associated protein-like (AIRAPL) is an evolutionary conserved regulator of cellular proteostasis in nematodes. A recent study demonstrated that AIRAPL-deficient mice develop a fully penetrant MPN-like disease [174]. Further analysis of the mechanisms underlying these findings demonstrated that AIRAPL promotes the ubiquitination and proteasome degradation of newly synthesized IGF-IR receptor polypeptides, which leads to apoptotic cell death. The lack of AIRAPL and its inhibitory effects on IGF-IR signaling and apoptosis, in reverse, lead to the development of MPN. This intriguing idea was further supported by: (1) targeting IGF-IR prevented MPN in AIRAPL-deficient mice as well as in mice carrying the Jak2V617F mutation; and (2) the expression of AIRAPL was found to be commonly lacking in human MPN samples [174].
Malignant lymphoma
The potential contribution of IGF-IR to the pathogenesis of the different subtypes of malignant lymphoma has not been sufficiently investigated. Some studies, however, have shown that IGF-IR is over-expressed in some types of lymphoma cells [22, 23, 175, 176]. Studies from our lab supported a role for IGF-IR in nucleophosmin-ALK-expressing (NPM-ALK+) T-cell lymphoma [22]. In this regard, IGF-IR and IGF-I are widely overexpressed in NPM-ALK+ T-cell lymphoma cell lines and in ALK+ lymphoma tumors from patients [22]. We were also able to identify novel reciprocal functional interactions between IGF-IR and NPM-ALK that were demonstrated through transfection experiments using wild-type and mutated constructs of IGF-IR and NPM-ALK [22, 65]. Transfection of an NPM-ALK+ T-cell lymphoma cell line with wild-type NPM-ALK significantly increased pIGF-IR, and transfection with wild-type IGF-IR induced a marked increase in pNPM-ALK [22, 65]. Transfection of non-functional mutants failed to induce similar effects. Our studies also showed that selective antagonism of IGF-IR with PPP decreased cell viability, induced apoptosis and G2/M-phase cell-cycle arrest, and decreased proliferation and colony formation of NPM-ALK+ T-cell lymphoma cells [22]. In a more recent work, similar findings were observed when GSK1838705, a small molecule IGF-IR/IR/ALK inhibitor, was utilized to treat a variety of different cancers with high expression of IGF-IR, including NPM-ALK+ T-cell lymphoma [177].
Furthermore, in our lab, we used NPM-ALK+ T-cell lymphoma as a cancer model to identify novel transcriptional and posttranscriptional mechanisms underlying the aberrant increase in IGF-IR expression (Figure 4). For instance, the transcription factors Ikaros isoform-1 (Ik-1) and myeloid zinc finger 1 (MZF1) are markedly decreased in this lymphoma. Importantly, we found that Ik-1 and MZF1 are capable of down-regulating IGF-IR gene expression by binding directly to its promoter and 5’ region [178]. As result of decreasing IGF-IR mRNA and protein expression, both Ik-1 and MZF1 also decreased the expression of phosphorylated IGF-IR, which subsequently decreased the phosphorylation of important downstream signaling proteins such as AKT and IRS-1. Ectopic expression of these transcription factors decreased lymphoma cell viability, proliferation, migration, and colony-forming potential. We also demonstrated that the decay of IGF-IR mRNA is significantly delayed in NPM-ALK+ T-cell lymphoma cell lines compared with normal T lymphocytes, suggesting a second possible mechanism by which IGF-IR is upregulated in these cells [178].
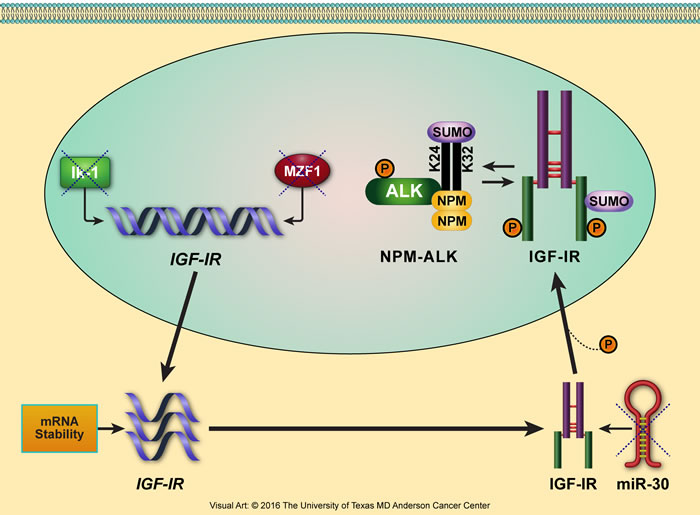
Figure 4: Proposed model illustrating the multilevel deregulatory mechanisms that lead to upregulation of IGF-IR expression in NPM-ALK+ T-cell lymphoma. We have been using NPM-ALK+ T-cell lymphoma as a cancer model to identify novel mechanisms underlying upregulation of IGF-IR expression. The levels of the transcription factors Ik-1 and MZF1 are significantly decreased in this lymphoma, preventing them from transcriptionally inhibiting IGF-IR gene expression and allowing IGF-IR mRNA to be transcribed. Furthermore, slower decay of IGF-IR mRNA increases its bioavailability for protein translation. In addition, microRNA-30a and microRNA-30d are also markedly decreased in NPM-ALK+ T-cell lymphoma (unpublished data). Lack of suppression of the IGF-IR-3’-UTR by these microRNAs leads to the posttranscriptional stabilization and upregulation of IGF-IR protein. IGF-IR protein is further stabilized by the posttranslational modification SUMOylation. SUMOylated IGF-IR can indirectly stabilize NPM-ALK, which is also capable of being SUMOylated [303].
To a lesser extent, a role of GH in regulating IGF-IR expression has been demonstrated in mouse T-cell EL4 lymphoma cells. It was shown that one of the consequences of overexpression of endogenous GH was an increase in the expression of IGF-I and IGF-IR, which mediated the protection of these lymphoma cells from apoptosis. As mentioned previously, GH regulates the secretion of IGF-I by the liver, although IGF-I concentrations can arise independently of GH. The EL4 lymphoma model may demonstrate a potential method of upregulation of IGF-IR via GH [175].
Our group has also demonstrated overexpression of IGF-IR in mantle cell lymphoma (MCL) cell lines and human lymphoma tissues [23]. The inhibition of IGF-IR by siRNA and PPP downregulated pIRS-1, pAKT, and pSTAT3 and induced caspase-3 cleavage in MCL. To our knowledge, these results were the first to report the expression of IRS-1 and pIRS-1 in MCL, which suggests that these proteins, through interactions with IGF-IR, could contribute to the survival of this aggressive lymphoma.
In Hodgkin lymphoma (HL), a recent study demonstrated that IGF-IR was overexpressed in 55% of the patients, and pIGF-IR was detectable in only 3 of the 16 IGF-IR-positive tumors that were analyzed for pIGF-IR expression [179]. In vitro experiments showed that the mitotic potential of HL cells is highly dependent on IGF-I, as treatment with the ligand resulted in increased phosphorylation of IGF-IR, AKT, and ERK in L428 and L1236 HL cells. Treatment with PPP was able to induce cell growth inhibition via G2/M-phase cell cycle arrest. In addition, positive IGF-IR status correlated with higher overall survival and 5-year progression-free survival rates compared with IGF-IR-negative patients, suggesting that positive IGF-IR status is in fact a prognostic marker for HL patients. This same phenomenon was also shown in a separate study, in which IGF-IR was associated with a better prognosis in patients with classical HL. However, this study also suggested that IGF-IR expression was closely associated with MET receptor expression, which allows another mode of therapeutic interference [180]. Despite the widely documented cancer-promoting effects of IGF-IR signaling, these two studies failed to provide an explanation for the better outcome of HL patients with IGF-IR expression in their tumors.
Diffuse large B-cell lymphoma (DLBCL) occurs in adults, and the standard chemotherapy treatment results in improvement in 60-70% of patients. Nonetheless, other treatment options are being sought for those in whom standard chemotherapy has failed. At least two IGF-IR inhibitors have been shown to efficiently induce cellular death in DLBCL in vitro: PPP and NVP-AEW541 [181]. As in other studies using PPP, DLBCL cells underwent apoptosis upon treatment with this inhibitor, proliferation was reduced, and cell growth was hindered through a G2/M-phase cell cycle arrest with complete elimination of cells in the G0/G1 phase of the cell cycle. Interestingly, however, PPP did not decrease the phosphorylation of IGF-IR at Tyr1131/Tyr1136 or its downstream targets AKT and ERK1/2, suggesting another possible mechanism of action in these cells. In contrast, NVP-AEW541 significantly inhibited phosphorylation of IGF-IR at Tyr1131/Tyr1136 and AKT, while ERK1/2 phosphorylation was less affected, suggesting that dual treatment with these inhibitors might provide more effective therapy for DLBCL [181].
PRECLINICAL AND CLINICAL EVALUATION OF IGF-IR INHIBITORS AND MECHANISMS OF RESISTANCE TO THESE INHIBITORS
One of the reasons why IGF-IR was initially considered a poor choice therapeutic option was the fact that it shares a high percentage of amino acid sequence identity with IR. However, more recently, there have been several approaches to target IGF-IR more specifically. Strategies to inhibit IGF-IR signaling in human cancers include: (1) monoclonal antibodies that directly block IGF-IR; (2) monoclonal antibodies that function by neutralizing the circulating ligands IGF-I or IGF-II, which prevents IGF-IR from being activated; and (3) small molecule inhibitors that antagonize the kinase activity of IGF-IR [182]. The anti-IGF-IR monoclonal antibodies sometimes result in insulin resistance, hyperinsulinemia, and hyperglycemia. Similar to IGF-IR monoclonal antibodies, the primary side effect of small molecule inhibitors is hyperglycemia, possibly through the inhibition of IR [183-187]. Table 1 lists the IGF-IR inhibitors that have been evaluated in preclinical studies. In addition, Table 2 lists the inhibitors that have been utilized in clinical trials, with different hematological neoplasms highlighted in RED.
Table 1: Inhibitors of IGF-IR that underwent pre-clinical evaluation.
Inhibitor |
Phase |
Tumor Type |
Manufacturer |
Type of inhibitor |
References |
A-928605 |
Pre-clinical |
Neuroblastoma |
Abbott |
Dual IGF-IR/IR tyrosine kinase inhibitor |
[283] |
ABDP |
Pre-clinical |
Colon cancer |
AstraZeneca |
Dual IGF-IR/IR tyrosine kinase inhibitor |
[284] |
AG-1024 |
Pre-clinical |
Breast cancer, colorectal cancer, glioma, hepatocellular carcinoma, mesothelioma, NSCLC, osteosarcoma, pancreatic cancer, prostate cancer |
Calbiochem EMD Biosciences |
Tyrosine kinase and autophosphorylation inhibitor |
|
BMS-536924 |
Pre-clinical |
AML, breast cancer, ovarian cancer |
Bristol-Myers Squibb |
ATP-competitive inhibitor |
|
BMS-554417 |
Pre-clinical |
Breast cancer, ovarian cancer |
Bristol-Myers Squibb |
Reversible ATP-competitive small molecule inhibitor |
[153] |
GSK1838705A |
Pre-clinical |
Breast cancer, cervical cancer, colon cancer, Ewing sarcoma, glioma, head and neck cancer, hepatocellular carcinoma, lung cancer, NPM-ALK+ T-cell lymphoma, ovarian cancer, PCM, pancreatic cancer, prostate cancer |
GlaxoSmithKline |
Triple IGF-IR/IR/ALK tyrosine kinase inhibitor |
|
GSK1904529A |
Pre-clinical |
Askin tumor, breast cancer, cervical cancer, colon cancer, Ewing sarcoma, head and neck cancer, lung cancer, NPM-ALK+ T-cell lymphoma, ovarian cancer, PCM, prostate cancer |
GlaxoSmithKline |
Dual IGF-IR/IR tyrosine kinase inhibitor |
[299] |
INSM-18 |
Pre-clinical |
Prostate cancer |
Insmed |
Dual IGF-IR/HER2 small molecule inhibitor |
[300] |
NVP-ADW742 |
Pre-clinical |
AML, Ewing sarcoma, medulloblastoma, NSCLC |
Novartis |
Reversible ATP-competitive small molecule inhibitor |
|
NVP-AEW541 |
Pre-clinical |
AML, breast cancer, colon cancer, endometrial cancer, esophageal cancer, Ewing sarcoma, gastric cancer, gastrointestinal stromal tumors, glioma, head and neck cancer, hepatocellular carcinoma, medulloblastoma, neuroblastoma, ovarian cancer, pancreatic cancer, PCM, pituitary tumors, prostate cancer, soft tissue sarcomas, synovial sarcoma |
Novartis |
Reversible ATP-competitive small molecule inhibitor |
|
Picropodophyllin (PPP; AXL1717) |
Pre-clinical |
Breast cancer, CML, colon cancer, Ewing sarcoma, glioblastoma, hepatocellular carcinoma, lung cancer, MCL, medulloblastoma, neuroblastoma, NPM-ALK+ T-cell lymphoma, osteosarcoma, ovarian cancer, pancreatic cancer, PCM, uveal melanoma |
Axelar |
Non-ATP-competitive small molecule inhibitor |
An example of the anti-IGF-IR blocking antibodies is AVE1642, which had encouraging effects in vitro and in vivo in animal models in different types of cancer including lung, hepatocellular carcinoma, PCM, Ewing sarcoma, and neuroblastoma, as well as against breast cancer metastasis [188-191]. Furthermore, it was well tolerated as a single agent or in combination with other chemotherapeutics in early clinical trials [192-194]. In addition, the IMC-A12 (cixutumumab) anti-IGF-IR monoclonal antibody achieved cancer growth inhibition in vitro and in xenograft models [195-198]. The efficacy of this antibody has since been evaluated in clinical trials, alone or in combination with other therapeutic agents [183-185, 199-202]. In addition to AVE1642 and cixutumumab, several other anti-IGF-IR monoclonal antibodies underwent preclinical and clinical trials evaluations, and are listed in Tables 1 and 2, respectively.
Table 2: IGF-IR inhibitors that were used in clinical trials.
Inhibitor |
Phase |
Tumor Type |
Manufacturer |
Type of Inhibitor |
Web links and references |
AMG 479 (Ganitumab) |
Phase I/II |
Breast cancer, colorectal cancer, Ewing sarcoma, lung cancer, lymphoma, melanoma, ovarian cancer, pancreatic cancer, refractory solid tumors, soft tissue sarcomas |
Amgen |
Monoclonal antibody (IgG1) |
|
AVE1642 |
Phase I/II |
Breast cancer, liver cancer, PCM |
Sanofi-Aventis |
Monoclonal antibody (IgG1) |
https://clinicaltrials.gov/ct2/results?term=AVE1642&Search=Search |
BIIB022 |
Phase I |
Hepatocellular carcinoma, NSCLC, refractory solid tumors |
Biogen |
Monoclonal antibody (IgG4) |
https://clinicaltrials.gov/ct2/results?term=BIIB022&Search=Search |
BMS-754807 |
Phase I/II |
Advanced or metastatic solid tumors, breast cancer, colorectal carcinoma, head and neck squamous cell carcinoma |
Bristol-Myers Squibb |
Dual IGF-IR/IR tyrosine kinase inhibitor-reversible ATP competitive inhibitor |
https://clinicaltrials.gov/ct2/results?term=BMS-754807&Search=Search |
CP-751,871 (Figitumumab) |
Phase I/II/III |
Advanced solid tumors, breast cancer, colorectal cancer, Ewing sarcoma, head and neck squamous cell carcinoma, lung cancer, PCM, prostate cancer |
Pfizer |
Monoclonal antibody (IgG2) |
https://clinicaltrials.gov/ct2/results?term=CP-751%2C871&Search=Search |
IMC-A12 (Cixutumumab) |
Phase I/II |
Adrenocortical carcinoma, brain stem neoplasms, breast cancer, ciliary body and choroid melanoma, colorectal cancer, esophageal carcinoma, Ewing sarcoma, head and neck squamous cell carcinoma, hepatocellular carcinoma, lung cancer, mesothelioma, neuroendocrine neoplasms, osteosarcoma, prostate cancer, recurrent or refractory solid tumors, soft tissue sarcomas, thymoma and thymic carcinoma |
ImClone |
Monoclonal antibody (IgG1) |
https://clinicaltrials.gov/ct2/results?term=IMC-A12&Search=Search |
MK-0646 (Dalotuzumab) |
Phase I/II |
Advanced solid tumors, breast cancer, colorectal cancer, lung cancer, neuroendocrine neoplasms, ovarian cancer, pancreatic cancer, PCM |
Merck |
Monoclonal antibody (IgG1) |
https://clinicaltrials.gov/ct2/results?term=MK-0646&Search=Search |
OSI-906 (Linsitinib) |
Phase I/II/III |
Advanced solid tumors, adrenocortical carcinoma, breast cancer, colorectal cancer, Ewing sarcoma, gastrointestinal stromal tumors, head and neck squamous cell carcinoma, hepatocellular carcinoma, lung cancer, ovarian cancer, pancreatic cancer, PCM, prostate cancer |
OSI Pharmaceuticals |
Reversible ATP-competitive small molecule inhibitor |
https://clinicaltrials.gov/ct2/results?term=OSI-906&Search=Search |
Picropodophyllin (PPP; AXL1717) |
Phase I/II |
Malignant astrocytomas, NSCLC |
Axelar |
Non-ATP-competitive small molecule inhibitor |
https://clinicaltrials.gov/ct2/results?term=Picropodophyllin+&Search=Search and [217] |
R1507 |
Phase I/II |
Breast cancer, Ewing sarcoma, NSCLC, osteosarcoma, soft tissue sarcomas |
Roche |
Monoclonal antibody (IgG1) |
https://clinicaltrials.gov/ct2/results?term=R1507&Search=Search |
SCH717454 (Robatumumab) |
Phase I/II |
Advanced solid tumors, colorectal cancer, Ewing sarcoma, neuroblastoma, osteosarcoma, pediatric solid tumors, soft tissue sarcomas |
Schering-Plough |
Monoclonal antibody (IgG1) |
https://clinicaltrials.gov/ct2/results?term=SCH717454&Search=Search |
XL-228 |
Phase I |
Advanced solid tumors, CML, lymphoma, PCM, Ph+ ALL |
Exelixis |
Dual IGF-IR/SRC small molecule inhibitor |
https://clinicaltrials.gov/ct2/results?term=XL-228&Search=Search |
The IGF-IR small molecule inhibitors compete for the binding site for ATP within the IGF-IR kinase domain. For example, in in vitro and in vivo preclinical models, PPP was shown to demonstrate pronounced inhibitory effects on cancer cells, including growth inhibition, cell cycle arrest, and apoptosis [21-23, 56, 64, 203-216]. Because of promising preclinical outcomes, the effects of PPP have also been explored in clinical trials [217]. NVP-AEW541 is another IGF-IR small molecule inhibitor that has been extensively tested in in vitro and in vivo preclinical studies in a variety of cancer cell types [56, 57, 61, 218-240]. Some of the small molecule inhibitors of IGF-IR that underwent preclinical evaluations are listed in Table 1, and, in addition, some of the ones tested in clinical trials are shown in Table 2.
Even when IGF-IR inhibitors were tolerated, significant number of patients did not exhibit improved outcomes. While further evaluations are required to characterize the mechanisms behind the shortcomings of these inhibitors, several studies underscored that an adequate number of IGF-IR receptors per cell or high levels of expression of IGF-IR protein or mRNA is required to achieve a robust response to the inhibitors [241-245]. Although an inherent low number of IGF-IR molecules per cancer cell may cause primary resistance to anti-IGF-IR antibodies or small molecule inhibitors, this factor appears to be more relevant to inducing secondary resistance when anti-IGF-IR antibodies are used. After the anti-IGF-IR antibody/IGF-IR complex undergoes endocytosis and subsequent degradation, a substantial reduction in the number of IGF-IR molecules on the surface of the cells typically occurs, which could cause secondary resistance after an initial response to the effects of IGF-IR antibodies [244]. The resistance to IGF-IR inhibitors can also stem from other members of the IGF system receptors including the IR, particularly IR-A and to a lesser extent IR-B [244, 246-248]. Moreover, the hybrid receptor IGF-IR/IR-A has also been suggested to induce significant resistance to IGF-IR inhibitors.
One of the widely studied mechanisms for cancer cell resistance to IGF-IR inhibition is the upregulation of AKT/mTOR signaling pathway. It was shown that inhibition of IGF-IR induces activation of AKT/mTOR, resulting in a feedback synthesis and activation of AKT as well as of epidermal growth factor receptor (EGFR) family and survivin. These proteins possess pronounced ability to overcome the effects of IGF-IR inhibitors and cause survival of the resistant cells [32, 234, 236, 241, 249-252]. Similarly, potentiation of ERK/MAPK signaling has been implicated in the resistance to IGF-IR inhibitors [32, 238, 244, 253]. Also, recent studies suggested that a bypass activation of SRC kinases family induces significant resistance to IGF-IR inhibitors in cancer cells [254]. Activated SRC acts through integrin α5β3, which leads to considerable resistance to IGF-IR inhibition [255]. Signaling through platelet-derived growth factor receptor-α (PDGFRα) has also been proposed to enhance the resistance to targeting IGF-IR [237, 243]. In support of this notion, PDGFRα is overexpressed and constitutively activated in cells resistant to IGF-IR inhibitors. Figure 5 illustrates some of the most commonly proposed mechanisms of resistance to IGF-IR inhibitors.
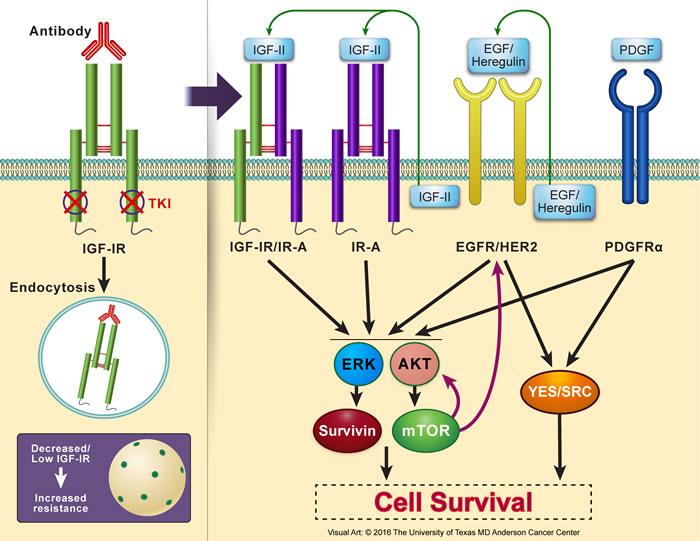
Figure 5: Resistance to IGF-IR inhibition in cancer cells. Several resistance mechanisms to IGF-IR inhibitors have been reported using in vitro and in vivo experimental models as well as proposed in clinical trials. Lack of abundant levels of IGF-IR protein expression or adequate number of IGF-IR receptor molecules per cell, which could be an inherent criterion of a specific type of cancer or acquired after initiation of IGF-IR inhibition therapy particularly with using IGF-IR antibodies that cause endocytosis and degradation of IGF-IR, is believed to be an important factor leading to resistance. Resistance to IGF-IR inhibition can be mediated through other members of the IGF system receptors that possess oncogenic potential such as IR-A or the hybrid receptor IGF-IR/IR-A. In addition, resistance can also be initiated through other non-IGF signaling systems. Examples of these include EGFR/HER2 and PDGFRα. Collectively, resistance systems bypass IGF-IR inhibition by providing alternate downstream activation of AKT and MAPK. These kinases further activate/sustain mTOR and survivin. mTOR, in return, contributes through a feedback loop to upregulation of EGFR/HER2 and AKT. EGFR/HER2 and PDGFRα can also surpass the effects of IGF-IR inhibition by activating the SRC kinases family. Additional studies are still required to further analyze the mechanisms that cause resistance to IGF-IR inhibitors, which is a very important limiting factor to the wide clinical utilization of these inhibitors to treat cancer patients. These studies may also shed light on mechanisms of resistance to other targeted therapeutics.
IGF-IR SIGNALING INDUCES Resistance to different CANCER THERAPEUTIC MODALITIES
The IGF-IR-related drug resistance in cancer does not only result from the utilization of IGF-IR inhibitors. In fact, IGF-IR signaling, by itself, can elicit substantial resistance to other cancer therapeutic modalities including chemotherapy and radiotherapy [256-263]. Moreover, IGF-IR has been shown to induce resistance to agents that selectively target oncogenic molecules other than IGF-IR. In a fashion reminiscent to the effects of EGFR family of kinases on inhibitors of IGF-IR, substantial evidence from several laboratories supported that IGF-IR signaling causes resistance to the blocking antibodies and small molecule inhibitors that interfere with the signaling of EGFR/HER2 kinases family [264-270]. This phenomenon has been reported in a variety of cancers including those of the lung, breast, and prostate. It is possible that heterodimerization between IGF-IR and EGFR/HER2 contributes to the resistance to EGFR inhibitors [271, 272]. In addition to resistance to the inhibition of EGFR, IGF-IR has been shown to induce significant resistance to inhibitors of several other cancer survival signaling including those functioning through modulation of the estrogen and androgen receptors (breast and prostate cancers, respectively), proteasome degradation (PCM), ALK kinase (NSCLC), ATM-related kinase (ATR; breast cancer), and the colony-stimulating factor-1 receptor (CSF-1R; gliomas) [273-279]. Although the mechanisms of IGF-IR-induced resistance are not completely defined, upregulation and bypass activation of survival-promoting proteins have been demonstrated. For example, a recent study showed that in breast cancer cells resistant to the effects of the anti-EGFR/HER2 antibody trastuzumab, IGF-IR induces resistance through the SRC/FAK/FoxM1 signaling [280]. Other studies also showed that IGF-IR-mediated upregulation of MAPK/ERK signaling could be another mechanism for drug resistance [275]. But one of the most frequently implicated modes of cancer cell resistance is the ability of IGF-IR to enhance PI3K/AKT/mTOR signaling, which could bypass the effects of the therapeutic agents. This mechanism has been demonstrated in different types of cancer cells including breast cancer, NSCLC, and head and neck squamous cell carcinoma cells, and by using different types of cancer antagonists including hormonal modulation, chemotherapeutic agents, EGFR small molecule inhibitors gefitinib and erlotinib, and AKT small molecule inhibitor AZD5363 [258, 264, 272, 275, 276]. IGF-IR-induced upregulation of PI3K/AKT/mTOR appears to induce its effects, at least in part, through upregulation of survivin expression [258, 267, 272].
Analyzing carefully the different routes of IGF-IR-induced drug resistance can possibly shed some light on how to intervene with novel therapeutics to overcome this setback. It is important to note that the mechanisms by which resistance to IGF-IR inhibitors evolve are largely similar to the mechanisms induced by IGF-IR against other therapeutic modalities. Therefore, it is not surprising that preclinical studies and clinical trials where combined targeting of IGF-IR and other survival promoting proteins that reciprocally interact with IGF-IR to cause drug resistance provided promising results implicating that such an approach could be more beneficial than using one agent alone [201, 258, 264, 272, 277, 278, 279, 280, 281, 282].
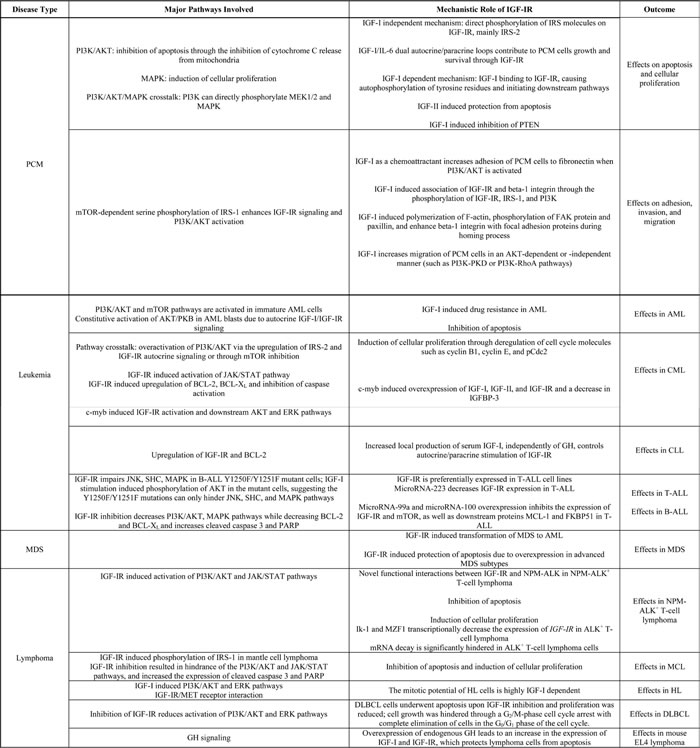
Table 3: Summary of mechanistic roles of IGF-IR in the pathogenesis of hematological malignancies.
Conclusions
In spite of the breakthroughs that have occurred in treating patients with hematological malignancies, important gaps in knowledge still exist in understanding the pathobiology of these aggressive neoplasms. Unraveling the mysteries underlying these gaps is expected to further facilitate the development of novel therapeutic strategies to tackle and eradicate these neoplasms. Although a relatively fewer studies have analyzed the role of IGF-IR in hematological neoplasms than in solid cancers, strong evidence suggests that this oncogenic receptor may become a useful target, alone or in combination, for treating hematological neoplasms.
Several in vitro and in vivo experimental models have provided links between IGF-IR signaling and downstream mediators that maintain the survival of PCM, leukemia, and lymphoma. Table 3 summarizes some of these mechanisms. Characterizing the effects of IGF-IR has led to the development of promising targeting approaches, including monoclonal antibodies and small molecule inhibitors that have shown potent antitumor activity in vitro and in animal models.
Although early evidence at least from some clinical trials suggested that targeting IGF-IR could represent a promising approach to treating cancer patients, the outcomes of these trials have not been as conclusive as the in vitro and animal research. We believe that one relevant explanation for this discrepancy is that the selection of patients included in the trials was mostly random and not rationally justified. The majority of these trials were phase I that included patients who have already been treated with several approaches that most likely have caused their cancers to undergo important molecular and biological modifications that resulted in resistance to more than one therapeutic modality. Instead, selection of patients to be treated by IGF-IR inhibitors, or similar targeted therapeutics, should be based on rigorous criteria such as proven biomarkers, stringent clinical characteristics, and/or histopathological and molecular assays. As an example, a higher success rate for IGF-IR inhibitors could have been achieved if an accurate estimation of the level of expression of IGF-IR and possibly the expression of pIGF-IR was first performed in the patients’ tumors prior to their enrollment in the trials. It is quite reasonable to assume that the presence of abundant expression of IGF-IR could positively impact the response to its inhibitors. Utilizing such strategies may improve the clinical outcomes of targeted therapeutics, including IGF-IR inhibitors, in cancer and, hopefully, in time eliminate this aggressive disease.
ACKNOWLEDGMENTS
We are grateful to Dawn Chalaire for outstanding assistance with the preparation of this paper, and to Jordan Pietz for exceptional help with the preparation of the figures. The authors apologize to the investigators whose contributions to this field could not be discussed or cited because of space limitations.
This work is supported by an R01CA151533 grant from the National Cancer Institute (NCI) to H.M.A., an R21CA170035 grant from the NCI to A.O.K., and by grant 14520720700 from Shanghai Scientific and Technological Innovation Project to P.S. The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the NCI or the National Institutes of Health.
COnflicts of INTERESTS
The authors declare no competing financial interests.
REFERENCES
1. Buchdunger E, Zimmermann J, Mett H, Meyer T, Muller M, Druker BJ, Lydon NB. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996; 56:100-104.
2. Deininger MW, Goldman JM, Lydon N, Melo JV. The tyrosine kinase inhibitor CGP57148B selectively inhibits the growth of BCR-ABL-positive cells. Blood. 1997; 90:3691-3698.
3. Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996; 2:561-566.
4. Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, Castaneda S, Cornelius LA, Das J, Doweyko AM, Fairchild C, Hunt JT, Inigo I et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004; 47:6658-6661.
5. Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A, Huntly B, Fabbro D, Fendrich G, Hall-Meyers E, Kung AL, Mestan J, Daley GQ et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005; 7:129-141.
6. Maloney DG, Liles TM, Czerwinski DK, Waldichuk C, Rosenberg J, Grillo-Lopez A, Levy R. Phase I clinical trial using escalating single-dose infusion of chimeric anti-CD20 monoclonal antibody (IDEC-C2B8) in patients with recurrent B-cell lymphoma. Blood. 1994; 84:2457-2466.
7. Kaleko M, Rutter WJ, Miller AD. Overexpression of the human insulinlike growth factor I receptor promotes ligand-dependent neoplastic transformation. Mol Cell Biol. 1990; 10:464-473.
8. Sell C, Dumenil G, Deveaud C, Miura M, Coppola D, DeAngelis T, Rubin R, Efstratiadis A, Baserga R. Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol Cell Biol. 1994; 14:3604-3612.
9. Resnicoff M, Abraham D, Yutanawiboonchai W, Rotman HL, Kajstura J, Rubin R, Zoltick P, Baserga R. The insulin-like growth factor I receptor protects tumor cells from apoptosis in vivo. Cancer Res. 1995; 55:2463-2469.
10. Sell C, Rubini M, Rubin R, Liu JP, Efstratiadis A, Baserga R. Simian virus 40 large tumor antigen is unable to transform mouse embryonic fibroblasts lacking type 1 insulin-like growth factor receptor. Proc Natl Acad Sci U S A. 1993; 90:11217-11221.
11. Himmelmann B, Terry C, Dey BR, Lopaczynski W, Nissley P. Anchorage-independent growth of fibroblasts that express a truncated IGF-I receptor. Biochem Biophys Res Commun. 2001; 286:472-477.
12. Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012; 12:159-169.
13. Ankrapp DP, Bevan DR. Insulin-like growth factor-I and human lung fibroblast-derived insulin-like growth factor-I stimulate the proliferation of human lung carcinoma cells in vitro. Cancer Res. 1993; 53:3399-3404.
14. De Leon DD, Wilson DM, Powers M, Rosenfeld RG. Effects of insulin-like growth factors (IGFs) and IGF receptor antibodies on the proliferation of human breast cancer cells. Growth Factors. 1992; 6:327-336.
15. Iwamura M, Sluss PM, Casamento JB, Cockett AT. Insulin-like growth factor I: action and receptor characterization in human prostate cancer cell lines. Prostate. 1993; 22:243-252.
16. Resnicoff M, Ambrose D, Coppola D, Rubin R. Insulin-like growth factor-1 and its receptor mediate the autocrine proliferation of human ovarian carcinoma cell lines. Lab Invest. 1993; 69:756-760.
17. Scotlandi K, Benini S, Sarti M, Serra M, Lollini PL, Maurici D, Picci P, Manara MC, Baldini N. Insulin-like growth factor I receptor-mediated circuit in Ewing’s sarcoma/peripheral neuroectodermal tumor: a possible therapeutic target. Cancer Res. 1996; 56:4570-4574.
18. Chapuis N, Tamburini J, Cornillet-Lefebvre P, Gillot L, Bardet V, Willems L, Park S, Green AS, Ifrah N, Dreyfus F, Mayeux P, Lacombe C, Bouscary D. Autocrine IGF-1/IGF-1R signaling is responsible for constitutive PI3K/Akt activation in acute myeloid leukemia: therapeutic value of neutralizing anti-IGF-1R antibody. Haematologica. 2010; 95:415-423.
19. Li W, Hyun T, Heller M, Yam A, Flechner L, Pierce JH, Rudikoff S. Activation of insulin-like growth factor I receptor signaling pathway is critical for mouse plasma cell tumor growth. Cancer Res. 2000; 60:3909-3915.
20. Oliveira MR, Ohnuma L, Bendit I, Dorlhiac-Lacer P, Giannella-Neto D. Interferon-alpha therapy increases type I insulin-like growth factor receptors expression on lymphoid cells from patients with chronic myelogenous leukemia. Leuk Res. 2001; 25:711-717.
21. Shi P, Chandra J, Sun X, Gergely M, Cortes JE, Garcia-Manero G, Arlinghaus RB, Lai R, Amin HM. Inhibition of IGF-IR tyrosine kinase induces apoptosis and cell cycle arrest in imatinib-resistant chronic myeloid leukaemia cells. J Cell Mol Med. 2010; 14:1777-1792.
22. Shi P, Lai R, Lin Q, Iqbal AS, Young LC, Kwak LW, Ford RJ, Amin HM. IGF-IR tyrosine kinase interacts with NPM-ALK oncogene to induce survival of T-cell ALK+ anaplastic large-cell lymphoma cells. Blood. 2009; 114:360-370.
23. Vishwamitra D, Shi P, Wilson D, Manshouri R, Vega F, Schlette EJ, Amin HM. Expression and effects of inhibition of type I insulin-like growth factor receptor tyrosine kinase in mantle cell lymphoma. Haematologica. 2011; 96:871-880.
24. Yaktapour N, Ubelhart R, Schuler J, Aumann K, Dierks C, Burger M, Pfeifer D, Jumaa H, Veelken H, Brummer T, Zirlik K. Insulin-like growth factor-1 receptor (IGF1R) as a novel target in chronic lymphocytic leukemia. Blood. 2013; 122:1621-1633.
25. Denley A, Cosgrove LJ, Booker GW, Wallace JC, Forbes BE. Molecular interactions of the IGF system. Cytokine Growth Factor Rev. 2005; 16:421-439.
26. Baxter RC. IGF binding proteins in cancer: mechanistic and clinical insights. Nat Rev Cancer. 2014; 14:329-341.
27. Ullrich A, Gray A, Tam AW, Yang-Feng T, Tsubokawa M, Collins C, Henzel W, Le Bon T, Kathuria S, Chen E, Jacobs S, Francke U, Ramachandran J et al. Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 1986; 5:2503-2512.
28. Tartari CJ, Gunby RH, Coluccia AM, Sottocornola R, Cimbro B, Scapozza L, Donella-Deana A, Pinna LA, Gambacorti-Passerini C. Characterization of some molecular mechanisms governing autoactivation of the catalytic domain of the anaplastic lymphoma kinase. J Biol Chem. 2008; 283:3743-3750.
29. Morris SW, Naeve C, Mathew P, James PL, Kirstein MN, Cui X, Witte DP. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene. 1997; 14:2175-2188.
30. Lee CC, Jia Y, Li N, Sun X, Ng K, Ambing E, Gao MY, Hua S, Chen C, Kim S, Michellys PY, Lesley SA, Harris JL et al. Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain. Biochem J. 2010; 430:425-437.
31. Chitnis MM, Yuen JS, Protheroe AS, Pollak M, Macaulay VM. The type 1 insulin-like growth factor receptor pathway. Clin Cancer Res. 2008; 14:6364-6370.
32. Bertrand FE, Steelman LS, Chappell WH, Abrams SL, Shelton JG, White ER, Ludwig DL, McCubrey JA. Synergy between an IGF-1R antibody and Raf/MEK/ERK and PI3K/Akt/mTOR pathway inhibitors in suppressing IGF-1R-mediated growth in hematopoietic cells. Leukemia. 2006; 20:1254-1260.
33. Zong CS, Chan J, Levy DE, Horvath C, Sadowski HB, Wang LH. Mechanism of STAT3 activation by insulin-like growth factor I receptor. J Biol Chem. 2000; 275:15099-15105.
34. Inagaki K, Tiulpakov A, Rubtsov P, Sverdlova P, Peterkova V, Yakar S, Terekhov S, LeRoith D. A familial insulin-like growth factor-I receptor mutant leads to short stature: clinical and biochemical characterization. J Clin Endocrinol Metab. 2007; 92:1542-1548.
35. Machwate M, Zerath E, Holy X, Pastoureau P, Marie PJ. Insulin-like growth factor-I increases trabecular bone formation and osteoblastic cell proliferation in unloaded rats. Endocrinology. 1994; 134:1031-1038.
36. Sadagurski M, Yakar S, Weingarten G, Holzenberger M, Rhodes CJ, Breitkreutz D, Leroith D, Wertheimer E. Insulin-like growth factor 1 receptor signaling regulates skin development and inhibits skin keratinocyte differentiation. Mol Cell Biol. 2006; 26:2675-2687.
37. Scavo LM, Karas M, Murray M, Leroith D. Insulin-like growth factor-I stimulates both cell growth and lipogenesis during differentiation of human mesenchymal stem cells into adipocytes. J Clin Endocrinol Metab. 2004; 89:3543-3553.
38. Yao W, Zhong J, Rosen CJ, Hock JM, Lee WH. Igf-I and postnatal growth of weaver mutant mice. Endocrine. 2005; 26:117-125.
39. Sun Z, Shushanov S, LeRoith D, Wood TL. Decreased IGF type 1 receptor signaling in mammary epithelium during pregnancy leads to reduced proliferation, alveolar differentiation, and expression of insulin receptor substrate (IRS)-1 and IRS-2. Endocrinology. 2011; 152:3233-3245.
40. Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell. 1993; 75:59-72.
41. Powell-Braxton L, Hollingshead P, Warburton C, Dowd M, Pitts-Meek S, Dalton D, Gillett N, Stewart TA. IGF-I is required for normal embryonic growth in mice. Genes Dev. 1993; 7:2609-2617.
42. Wu Y, Sun H, Yakar S, LeRoith D. Elevated levels of insulin-like growth factor (IGF)-I in serum rescue the severe growth retardation of IGF-I null mice. Endocrinology. 2009; 150:4395-4403.
43. Zhao G, Monier-Faugere MC, Langub MC, Geng Z, Nakayama T, Pike JW, Chernausek SD, Rosen CJ, Donahue LR, Malluche HH, Fagin JA, Clemens TL. Targeted overexpression of insulin-like growth factor I to osteoblasts of transgenic mice: increased trabecular bone volume without increased osteoblast proliferation. Endocrinology. 2000; 141:2674-2682.
44. Zhang D, Samani AA, Brodt P. The role of the IGF-I receptor in the regulation of matrix metalloproteinases, tumor invasion and metastasis. Horm Metab Res. 2003; 35:802-808.
45. Jones RA, Campbell CI, Petrik JJ, Moorehead RA. Characterization of a novel primary mammary tumor cell line reveals that cyclin D1 is regulated by the type I insulin-like growth factor receptor. Mol Cancer Res. 2008; 6:819-828.
46. Rubini M, Hongo A, D’Ambrosio C, Baserga R. The IGF-I receptor in mitogenesis and transformation of mouse embryo cells: role of receptor number. Exp Cell Res. 1997; 230:284-292.
47. Flossmann-Kast BB, Jehle PM, Hoeflich A, Adler G, Lutz MP. Src stimulates insulin-like growth factor I (IGF-I)-dependent cell proliferation by increasing IGF-I receptor number in human pancreatic carcinoma cells. Cancer Res. 1998; 58:3551-3554.
48. Lann D, LeRoith D. The role of endocrine insulin-like growth factor-I and insulin in breast cancer. J Mammary Gland Biol Neoplasia. 2008; 13:371-379.
49. Cannata D, Lann D, Wu Y, Elis S, Sun H, Yakar S, Lazzarino DA, Wood TL, LeRoith D. Elevated circulation IGF-I promotes mammary gland development and proliferation. Endocrinology. 2010; 151:5751-5761.
50. Sayeed A, Alam N, Trerotola M, Languino LR. Insulin-like growth factor 1 stimulation of androgen receptor activity requires beta(1A) integrins. J Cell Physiol. 2012; 227:751-758.
51. Wu JD, Haugk K, Woodke L, Nelson P, Coleman I, Plymate SR. Interaction of IGF signaling and the androgen receptor in prostate cancer progression. J Cell Biochem. 2006; 99:392-401.
52. Freier S, Weiss O, Eran M, Flyvbjerg A, Dahan R, Nephesh I, Safra T, Shiloni E, Raz I. Expression of the insulin-like growth factors and their receptors in adenocarcinoma of the colon. Gut. 1999; 44:704-708.
53. Wolpin BM, Meyerhardt JA, Chan AT, Ng K, Chan JA, Wu K, Pollak MN, Giovannucci EL, Fuchs CS. Insulin, the insulin-like growth factor axis, and mortality in patients with nonmetastatic colorectal cancer. J Clin Oncol. 2009; 27:176-185.
54. Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Shringarpure R, Akiyama M, Hideshima T, Chauhan D, Joseph M, Libermann TA, Garcia-Echeverria C, Pearson MA, Hofmann F et al. Inhibition of the insulin-like growth factor receptor-1 tyrosine kinase activity as a therapeutic strategy for multiple myeloma, other hematologic malignancies, and solid tumors. Cancer Cell. 2004; 5:221-230.
55. Chng WJ, Gualberto A, Fonseca R. IGF-1R is overexpressed in poor-prognostic subtypes of multiple myeloma. Leukemia. 2006; 20:174-176.
56. Tazzari PL, Tabellini G, Bortul R, Papa V, Evangelisti C, Grafone T, Martinelli G, McCubrey JA, Martelli AM. The insulin-like growth factor-I receptor kinase inhibitor NVP-AEW541 induces apoptosis in acute myeloid leukemia cells exhibiting autocrine insulin-like growth factor-I secretion. Leukemia. 2007; 21:886-896.
57. Doepfner KT, Spertini O, Arcaro A. Autocrine insulin-like growth factor-I signaling promotes growth and survival of human acute myeloid leukemia cells via the phosphoinositide 3-kinase/Akt pathway. Leukemia. 2007; 21:1921-1930.
58. Tamburini J, Chapuis N, Bardet V, Park S, Sujobert P, Willems L, Ifrah N, Dreyfus F, Mayeux P, Lacombe C, Bouscary D. Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: rationale for therapeutic inhibition of both pathways. Blood. 2008; 111:379-382.
59. Wahner Hendrickson AE, Haluska P, Schneider PA, Loegering DA, Peterson KL, Attar R, Smith BD, Erlichman C, Gottardis M, Karp JE, Carboni JM, Kaufmann SH. Expression of insulin receptor isoform A and insulin-like growth factor-1 receptor in human acute myelogenous leukemia: effect of the dual-receptor inhibitor BMS-536924 in vitro. Cancer Res. 2009; 69:7635-7643.
60. He Y, Zhang J, Zheng J, Du W, Xiao H, Liu W, Li X, Chen X, Yang L, Huang S. The insulin-like growth factor-1 receptor kinase inhibitor, NVP-ADW742, suppresses survival and resistance to chemotherapy in acute myeloid leukemia cells. Oncol Res. 2010; 19:35-43.
61. Weisberg E, Nonami A, Chen Z, Nelson E, Chen Y, Liu F, Cho H, Zhang J, Sattler M, Mitsiades C, Wong KK, Liu Q, Gray NS et al. Upregulation of IGF1R by mutant RAS in leukemia and potentiation of RAS signaling inhibitors by small-molecule inhibition of IGF1R. Clin Cancer Res. 2014; 20:5483-5495.
62. Whelan JT, Ludwig DL, Bertrand FE. HoxA9 induces insulin-like growth factor-1 receptor expression in B-lineage acute lymphoblastic leukemia. Leukemia. 2008; 22:1161-1169.
63. Medyouf H, Gusscott S, Wang H, Tseng JC, Wai C, Nemirovsky O, Trumpp A, Pflumio F, Carboni J, Gottardis M, Pollak M, Kung AL, Aster JC et al. High-level IGF1R expression is required for leukemia-initiating cell activity in T-ALL and is supported by Notch signaling. J Exp Med. 2011; 208:1809-1822.
64. Huang Z, Fang Z, Zhen H, Zhou L, Amin HM, Shi P. Inhibition of type I insulin-like growth factor receptor tyrosine kinase by picropodophyllin induces apoptosis and cell cycle arrest in T lymphoblastic leukemia/lymphoma. Leuk Lymphoma. 2014; 55:1876-1883.
65. Shi B, Vishwamitra D, Granda JG, Whitton T, Shi P, Amin HM. Molecular and functional characterizations of the association and interactions between nucleophosmin-anaplastic lymphoma kinase and type I insulin-like growth factor receptor. Neoplasia. 2013; 15:669-683.
66. Kornfeld S. Structure and function of the mannose 6-phosphate/insulinlike growth factor II receptors. Annu Rev Biochem. 1992; 61:307-330.
67. Brown J, Delaine C, Zaccheo OJ, Siebold C, Gilbert RJ, van Boxel G, Denley A, Wallace JC, Hassan AB, Forbes BE, Jones EY. Structure and functional analysis of the IGF-II/IGF2R interaction. EMBO J. 2008; 27:265-276.
68. Brown J, Jones EY, Forbes BE. Keeping IGF-II under control: lessons from the IGF-II-IGF2R crystal structure. Trends Biochem Sci. 2009; 34:612-619.
69. Bhattacharyya N, Tao J, Klein EA, Banerjee S. Alterations of transforming growth factor beta receptor II, insulin growth factor receptor II genes in microsatellite unstable prostate carcinomas. Oncol Rep. 2004; 11:231-236.
70. Seitz S, Wassmuth P, Plaschke J, Schackert HK, Karsten U, Santibanez-Koref MF, Schlag PM, Scherneck S. Identification of microsatellite instability and mismatch repair gene mutations in breast cancer cell lines. Genes Chromosomes Cancer. 2003; 37:29-35.
71. Caligo MA, Ghimenti C, Sensi E, Marchetti A, Bertacca G, Giulianotti PG, Fornaciari G, Bevilacqua G. Microsatellite alterations and K-ras, TGFbetaRII, IGFRII and bax mutations in sporadic cancers of the gastrointestinal tract. Oncol Rep. 2000; 7:1371-1375.
72. De Souza AT, Hankins GR, Washington MK, Fine RL, Orton TC, Jirtle RL. Frequent loss of heterozygosity on 6q at the mannose 6-phosphate/insulin-like growth factor II receptor locus in human hepatocellular tumors. Oncogene. 1995; 10:1725-1729.
73. Rey JM, Theillet C, Brouillet JP, Rochefort H. Stable amino-acid sequence of the mannose-6-phosphate/insulin-like growth-factor-II receptor in ovarian carcinomas with loss of heterozygosity and in breast-cancer cell lines. Int J Cancer. 2000; 85:466-473.
74. Kuhlmann JD, Schwarzenbach H, Otterbach F, Heubner M, Wimberger P, Worm KH, Kimmig R, Kasimir-Bauer S. Loss of heterozygosity proximal to the M6P/IGF2R locus is predictive for the presence of disseminated tumor cells in the bone marrow of ovarian cancer patients before and after chemotherapy. Genes Chromosomes Cancer. 2011; 50:598-605.
75. Hu CK, McCall S, Madden J, Huang H, Clough R, Jirtle RL, Anscher MS. Loss of heterozygosity of M6P/IGF2R gene is an early event in the development of prostate cancer. Prostate Cancer Prostatic Dis. 2006; 9:62-67.
76. Jamieson TA, Brizel DM, Killian JK, Oka Y, Jang HS, Fu X, Clough RW, Vollmer RT, Anscher MS, Jirtle RL. M6P/IGF2R loss of heterozygosity in head and neck cancer associated with poor patient prognosis. BMC Cancer. 2003; 3:4.
77. Kong FM, Anscher MS, Washington MK, Killian JK, Jirtle RL. M6P/IGF2R is mutated in squamous cell carcinoma of the lung. Oncogene. 2000; 19:1572-1578.
78. Byrd JC, Devi GR, de Souza AT, Jirtle RL, MacDonald RG. Disruption of ligand binding to the insulin-like growth factor II/mannose 6-phosphate receptor by cancer-associated missense mutations. J Biol Chem. 1999; 274:24408-24416.
79. Rinderknecht E, Humbel RE. The amino acid sequence of human insulin-like growth factor I and its structural homology with proinsulin. J Biol Chem. 1978; 253:2769-2776.
80. Yakar S, Wu Y, Setser J, Rosen CJ. The role of circulating IGF-I: lessons from human and animal models. Endocrine. 2002; 19:239-248.
81. Wang J, Zhou J, Cheng CM, Kopchick JJ, Bondy CA. Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J Endocrinol. 2004; 180:247-255.
82. Chen W, Wang S, Tian T, Bain J, Hu Z, Xu Y, Dong J, Chen F, Wang X, Shen H. Phenotypes and genotypes of insulin-like growth factor 1, IGF-binding protein-3 and cancer risk: evidence from 96 studies. Eur J Hum Genet. 2009; 17:1668-1675.
83. Renehan AG, Zwahlen M, Minder C, O’Dwayer ST, Shalet SM, Egger M. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: sytetmatic review and meta-regression analysis. Lancet. 2004; 363:1346-1353.
84. Shariat SF, Lamb DJ, Kattan MW, Nguyen C, Kim J, Beck J, Wheeler TM, Slawin KM. Association of preoperative plasma levels of insulin-like growth factor I and insulin-like growth factor binding proteins-2 and -3 with prostate cancer invasion, progression, and metastasis. J Clin Oncol. 2002; 20:833-841.
85. Wu X, Tortolero-Luna G, Zhao H, Phatak D, Spitz MR, Follen M. Serum levels of insulin-like growth factor I and risk of squamous intraepithelial lesions of the cervix. Clin Cancer Res. 2003; 9:3356-3361.
86. Gao Y, Katki H, Graubard B, Pollak M, Martin M, Tao Y, Schoen RE, Church T, Hayes RB, Greene MH, Berndt SI. Serum IGF1, IGF2 and IGFBP3 and risk of advanced colorectal adenoma. Int J Cancer. 2012; 131:E105-113.
87. Takahara K, Tearle H, Ghaffari M, Gleave ME, Pollak M, Cox ME. Human prostate cancer xenografts in lit/lit mice exhibit reduced growth and androgen-independent progression. Prostate. 2011; 71:525-537.
88. DiGiovanni J, Kiguchi K, Frijhoff A, Wilker E, Bol DK, Beltran L, Moats S, Ramirez A, Jorcano J, Conti C. Deregulated expression of insulin-like growth factor 1 in prostate epithelium leads to neoplasia in transgenic mice. Proc Natl Acad Sci U S A. 2000; 97:3455-3460.
89. Lee AV, Jackson JG, Gooch JL, Hilsenbeck SG, Coronado-Heinsohn E, Osborne CK, Yee D. Enhancement of insulin-like growth factor signaling in human breast cancer: estrogen regulation of insulin receptor substrate-1 expression in vitro and in vivo. Mol Endocrinol. 1999; 13:787-796.
90. DeChiara TM, Efstratiadis A, Robertson EJ. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature. 1990; 345:78-80.
91. Daughaday WH, Rotwein P. Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr Rev. 1989; 10:68-91.
92. Zapf J, Froesch ER. Insulin-like growth factors/somatomedins: structure, secretion, biological actions and physiological role. Horm Res. 1986; 24:121-130.
93. Soares MB, Turken A, Ishii D, Mills L, Episkopou V, Cotter S, Zeitlin S, Efstratiadis A. Rat insulin-like growth factor II gene. A single gene with two promoters expressing a multitranscript family. J Mol Biol. 1986; 192:737-752.
94. Constancia M, Hemberger M, Hughes J, Dean W, Ferguson-Smith A, Fundele R, Stewart F, Kelsey G, Fowden A, Sibley C, Reik W. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature. 2002; 417:945-948.
95. Tollefsen SE, Sadow JL, Rotwein P. Coordinate expression of insulin-like growth factor II and its receptor during muscle differentiation. Proc Natl Acad Sci U S A. 1989; 86:1543-1547.
96. Back K, Brannmark C, Stralfors P, Arnqvist HJ. Differential effects of IGF-I, IGF-II and insulin in human preadipocytes and adipocytes—role of insulin and IGF-I receptors. Mol Cell Endocrinol. 2011; 339:130-135.
97. Burvin R, LeRoith D, Harel H, Zloczower M, Marbach M, Karnieli E. The effect of acute insulin-like growth factor-II administration on glucose metabolism in the rat. Growth Horm IGF Res. 1998; 8:205-210.
98. Gude MF, Frystyk J, Flyvbjerg A, Bruun JM, Richelsen B, Pedersen SB. The production and regulation of IGF and IGFBPs in human adipose tissue cultures. Growth Horm IGF Res. 2012; 22:200-205.
99. Han VK, D’Ercole AJ, Lund PK. Cellular localization of somatomedin (insulin-like growth factor) messenger RNA in the human fetus. Science. 1987; 236:193-197.
100. Hill DJ, Strutt B, Arany E, Zaina S, Coukell S, Graham CF. Increased and persistent circulating insulin-like growth factor II in neonatal transgenic mice suppresses developmental apoptosis in the pancreatic islets. Endocrinology. 2000; 141:1151-1157.
101. Liu L, Greenberg S, Russell SM, Nicoll CS. Effects of insulin-like growth factors I and II on growth and differentiation of transplanted rat embryos and fetal tissues. Endocrinology. 1989; 124:3077-3082.
102. Piecewicz SM, Pandey A, Roy B, Xiang SH, Zetter BR, Sengupta S. Insulin-like growth factors promote vasculogenesis in embryonic stem cells. PLoS One. 2012; 7:e32191.
103. Schwartz GN, Hudgins WR, Perdue JF. Glycosylated insulin-like growth factor II promoted expansion of granulocyte-macrophage colony-forming cells in serum-deprived liquid cultures of human peripheral blood cells. Exp Hematol. 1993; 21:1447-1454.
104. Siddals KW, Westwood M, Gibson JM, White A. IGF-binding protein-1 inhibits IGF effects on adipocyte function: implications for insulin-like actions at the adipocyte. J Endocrinol. 2002; 174:289-297.
105. Morison IM, Becroft DM, Taniguchi T, Woods CG, Reeve AE. Somatic overgrowth associated with overexpression of insulin-like growth factor II. Nat Med. 1996; 2:311-316.
106. Ravenel JD, Broman KW, Perlman EJ, Niemitz EL, Jayawardena TM, Bell DW, Haber DA, Uejima H, Feinberg AP. Loss of imprinting of insulin-like growth factor-II (IGF2) gene in distinguishing specific biologic subtypes of Wilms tumor. J Natl Cancer Inst. 2001; 93:1698-1703.
107. Lui JC, Baron J. Evidence that Igf2 down-regulation in postnatal tissues and up-regulation in malignancies is driven by transcription factor E2f3. Proc Natl Acad Sci U S A. 2013; 110:6181-6186.
108. Li J, Neumann I, Volkmer I, Staege MS. Down-regulation of achaete-scute complex homolog 1 (ASCL1) in neuroblastoma cells induces up-regulation of insulin-like growth factor 2 (IGF2). Mol Biol Rep. 2011; 38:1515-1521.
109. von Horn H, Ekstrom C, Ellis E, Olivecrona H, Einarsson C, Tally M, Ekstrom TJ. GH is a regulator of IGF2 promoter-specific transcription in human liver. J Endocrinol. 2002; 172:457-465.
110. Uchida K, Kondo M, Takeda S, Osada H, Takahashi T, Nakao A, Takahashi T. Altered transcriptional regulation of the insulin-like growth factor 2 gene in human hepatocellular carcinoma. Mol Carcinog. 1997; 18:193-198.
111. Caricasole A, Ward A. Transactivation of mouse insulin-like growth factor II (IGF-II) gene promoters by the AP-1 complex. Nucleic Acids Res. 1993; 21:1873-1879.
112. Yun K, Fidler AE, Eccles MR, Reeve AE. Insulin-like growth factor II and WT1 transcript localization in human fetal kidney and Wilms’ tumor. Cancer Res. 1993; 53:5166-5171.
113. Feng Y, Zhu Z, Xiao X, Choudhry V, Barrett JC, Dimitrov DS. Novel human monoclonal antibodies to insulin-like growth factor (IGF)-II that potently inhibit the IGF receptor type I signal transduction function. Mol Cancer Ther. 2006; 5:114-120.
114. Bates P, Fisher R, Ward A, Richardson L, Hill DJ, Graham CF. Mammary cancer in transgenic mice expressing insulin-like growth factor II (IGF-II). Br J Cancer. 1995; 72:1189-1193.
115. Boulle N, Logie A, Gicquel C, Perin L, Le Bouc Y. Increased levels of insulin-like growth factor II (IGF-II) and IGF-binding protein-2 are associated with malignancy in sporadic adrenocortical tumors. J Clin Endocrinol Metab. 1998; 83:1713-1720.
116. Ilvesmaki V, Kahri AI, Miettinen PJ, Voutilainen R. Insulin-like growth factors (IGFs) and their receptors in adrenal tumors: high IGF-II expression in functional adrenocortical carcinomas. J Clin Endocrinol Metab. 1993; 77:852-858.
117. Miyamoto S, Nakamura M, Shitara K, Nakamura K, Ohki Y, Ishii G, Goya M, Kodama K, Sangai T, Maeda H, Shi-Chuang Z, Chiba T, Ochiai A. Blockade of paracrine supply of insulin-like growth factors using neutralizing antibodies suppresses the liver metastasis of human colorectal cancers. Clin Cancer Res. 2005; 11:3494-3502.
118. Moorehead RA, Sanchez OH, Baldwin RM, Khokha R. Transgenic overexpression of IGF-II induces spontaneous lung tumors: a model for human lung adenocarcinoma. Oncogene. 2003; 22:853-857.
119. Reeve AE, Eccles MR, Wilkins RJ, Bell GI, Millow LJ. Expression of insulin-like growth factor-II transcripts in Wilms’ tumour. Nature. 1985; 317:258-260.
120. Sohda T, Iwata K, Soejima H, Kamimura S, Shijo H, Yun K. In situ detection of insulin-like growth factor II (IGF2) and H19 gene expression in hepatocellular carcinoma. J Hum Genet. 1998; 43:49-53.
121. Rogler CE, Yang D, Rossetti L, Donohoe J, Alt E, Chang CJ, Rosenfeld R, Neely K, Hintz R. Altered body composition and increased frequency of diverse malignancies in insulin-like growth factor-II transgenic mice. J Biol Chem. 1994; 269:13779-13784.
122. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011; 364:1046-1060.
123. Eleutherakis-Papaiakovou V, Karali M, Kokkonouzis I, Tiliakos I, Dimopoulos MA. Bone marrow angiogenesis and progression in multiple myeloma: clinical significance and therapeutic approach. Leuk Lymphoma. 2003; 44:937-948.
124. Ribatti D, Vacca A, Nico B, Quondamatteo F, Ria R, Minischetti M, Marzullo A, Herken R, Roncali L, Dammacco F. Bone marrow angiogenesis and mast cell density increase simultaneously with progression of human multiple myeloma. Br J Cancer. 1999; 79:451-455.
125. Vacca A, Ribatti D, Roncali L, Ranieri G, Serio G, Silvestris F, Dammacco F. Bone marrow angiogenesis and progression in multiple myeloma. Br J Haematol. 1994; 87:503-508.
126. Chauhan D, Kharbanda S, Ogata A, Urashima M, Teoh G, Robertson M, Kufe DW, Anderson KC. Interleukin-6 inhibits Fas-induced apoptosis and stress-activated protein kinase activation in multiple myeloma cells. Blood. 1997; 89:227-234.
127. Dankbar B, Padro T, Leo R, Feldmann B, Kropff M, Mesters RM, Serve H, Berdel WE, Kienast J. Vascular endothelial growth factor and interleukin-6 in paracrine tumor-stromal cell interactions in multiple myeloma. Blood. 2000; 95:2630-2636.
128. Villunger A, Egle A, Kos M, Hittmair A, Maly K, Greil R. Constituents of autocrine IL-6 loops in myeloma cell lines and their targeting for suppression of neoplastic growth by antibody strategies. Int J Cancer. 1996; 65:498-505.
129. Tsunenari T, Koishihara Y, Nakamura A, Moriya M, Ohkawa H, Goto H, Shimazaki C, Nakagawa M, Ohsugi Y, Kishimoto T, Akamatsu K. New xenograft model of multiple myeloma and efficacy of a humanized antibody against human interleukin-6 receptor. Blood. 1997; 90:2437-2444.
130. Jelinek DF, Witzig TE, Arendt BK. A role for insulin-like growth factor in the regulation of IL-6-responsive human myeloma cell line growth. J Immunol. 1997; 159:487-496.
131. Georgii-Hemming P, Wiklund HJ, Ljunggren O, Nilsson K. Insulin-like growth factor I is a growth and survival factor in human multiple myeloma cell lines. Blood. 1996; 88:2250-2258.
132. Sprynski AC, Hose D, Caillot L, Reme T, Shaughnessy JD, Jr., Barlogie B, Seckinger A, Moreaux J, Hundemer M, Jourdan M, Meissner T, Jauch A, Mahtouk K et al. The role of IGF-1 as a major growth factor for myeloma cell lines and the prognostic relevance of the expression of its receptor. Blood. 2009; 113:4614-4626.
133. Ferlin M, Noraz N, Hertogh C, Brochier J, Taylor N, Klein B. Insulin-like growth factor induces the survival and proliferation of myeloma cells through an interleukin-6-independent transduction pathway. Br J Haematol. 2000; 111:626-634.
134. DeAngelis T, Chen J, Wu A, Prisco M, Baserga R. Transformation by the simian virus 40 T antigen is regulated by IGF-I receptor and IRS-1 signaling. Oncogene. 2006; 25:32-42.
135. Richards RG, DiAugustine RP, Petrusz P, Clark CC, Sebastian J. Estradiol stimulates tyrosine phosphorylation of the insulin-like growth factor-1 receptor and insulin receptor substrate-1 in the uterus. Proc Natl Acad Sci USA. 1996; 93:12002-12007.
136. Abroun S, Ishikawa H, Tsuyama N, Liu S, Li FJ, Otsuyama K, Zheng X, Obata M, Kawano MM. Receptor synergy of interleukin-6 (IL-6) and insulin-like growth factor-I in myeloma cells that highly express IL-6 receptor alpha [corrected]. Blood. 2004; 103:2291-2298.
137. Tu Y, Gardner A, Lichtenstein A. The phosphatidylinositol 3-kinase/AKT pathway in multiple myeloma plasma cells: roles in cytokine-dependent survival and proliferative responses. Cancer Res. 2000; 60:6763-6770.
138. Ge NL, Rudikoff S. Insulin-like growth factor I is a dual effector of multiple myeloma cell growth. Blood. 2000; 96:2856-2861.
139. Choi Y, Zhang J, Murga C, Yu H, Koller E, Monia BP, Gutkind JS, Li W. PTEN, but not SHIP and SHIP2, suppresses the PI3K/Akt pathway and induces growth inhibition and apoptosis of myeloma cells. Oncogene. 2002; 21:5289-5300.
140. Hyun T, Yam A, Pece S, Xie X, Zhang J, Miki T, Gutkind JS, Li W. Loss of PTEN expression leading to high Akt activation in human multiple myelomas. Blood. 2000; 96:3560-3568.
141. Qiang YW, Kopantzev E, Rudikoff S. Insulinlike growth factor-I signaling in multiple myeloma: downstream elements, functional correlates, and pathway cross-talk. Blood. 2002; 99:4138-4146.
142. Shi Y, Yan H, Frost P, Gera J, Lichtenstein A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol Cancer Ther. 2005; 4:1533-1540.
143. Stromberg T, Ekman S, Girnita L, Dimberg LY, Larsson O, Axelson M, Lennartsson J, Hellman U, Carlson K, Osterborg A, Vanderkerken K, Nilsson K, Jernberg-Wiklund H. IGF-1 receptor tyrosine kinase inhibition by the cyclolignan PPP induces G2/M-phase accumulation and apoptosis in multiple myeloma cells. Blood. 2006; 107:669-678.
144. Stromberg T, Dimberg A, Hammarberg A, Carlson K, Osterborg A, Nilsson K, Jernberg-Wiklund H. Rapamycin sensitizes multiple myeloma cells to apoptosis induced by dexamethasone. Blood. 2004; 103:3138-3147.
145. Xu F, Gardner A, Tu Y, Michl P, Prager D, Lichtenstein A. Multiple myeloma cells are protected against dexamethasone-induced apoptosis by insulin-like growth factors. Br J Haematol. 1997; 97:429-440.
146. Tai YT, Podar K, Catley L, Tseng YH, Akiyama M, Shringarpure R, Burger R, Hideshima T, Chauhan D, Mitsiades N, Richardson P, Munshi NC, Kahn CR et al. Insulin-like growth factor-1 induces adhesion and migration in human multiple myeloma cells via activation of beta1-integrin and phosphatidylinositol 3’-kinase/AKT signaling. Cancer Res. 2003; 63:5850-5858.
147. Salani B, Briatore L, Contini P, Passalacqua M, Melloni E, Paggi A, Cordera R, Maggi D. IGF-I induced rapid recruitment of integrin beta1 to lipid rafts is Caveolin-1 dependent. Biochem Biophys Res Commun. 2009; 380:489-492.
148. Menu E, Kooijman R, Van Valckenborgh E, Asosingh K, Bakkus M, Van Camp B, Vanderkerken K. Specific roles for the PI3K and the MEK-ERK pathway in IGF-1-stimulated chemotaxis, VEGF secretion and proliferation of multiple myeloma cells: study in the 5T33MM model. Br J Cancer. 2004; 90:1076-1083.
149. Qiang YW, Yao L, Tosato G, Rudikoff S. Insulin-like growth factor I induces migration and invasion of human multiple myeloma cells. Blood. 2004; 103:301-308.
150. Yasui H, Hideshima T, Richardson PG, Anderson KC. Novel therapeutic strategies targeting growth factor signalling cascades in multiple myeloma. Br J Haematol. 2006; 132:385-397.
151. Xu Q, Simpson SE, Scialla TJ, Bagg A, Carroll M. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood. 2003; 102:972-980.
152. Grandage VL, Gale RE, Linch DC, Khwaja A. PI3-kinase/Akt is constitutively active in primary acute myeloid leukaemia cells and regulates survival and chemoresistance via NF-kappaB, Mapkinase and p53 pathways. Leukemia. 2005; 19:586-594.
153. Haluska P, Carboni JM, Loegering DA, Lee FY, Wittman M, Saulnier MG, Frennesson DB, Kalli KR, Conover CA, Attar RM, Kaufmann SH, Gottardis M, Erlichman C. In vitro and in vivo antitumor effects of the dual insulin-like growth factor-I/insulin receptor inhibitor, BMS-554417. Cancer Res. 2006; 66:362-371.
154. Deschler B, Lubbert M. Acute myeloid leukemia: epidemiology and etiology. Cancer. 2006; 107:2099-2107.
155. Bouscary D, De Vos J, Guesnu M, Jondeau K, Viguier F, Melle J, Picard F, Dreyfus F, Fontenay-Roupie M. Fas/Apo-1 (CD95) expression and apoptosis in patients with myelodysplastic syndromes. Leukemia. 1997; 11:839-845.
156. Kurotaki H, Tsushima Y, Nagai K, Yagihashi S. Apoptosis, bcl-2 expression and p53 accumulation in myelodysplastic syndrome, myelodysplastic-syndrome-derived acute myelogenous leukemia and de novo acute myelogenous leukemia. Acta Haematol. 2000; 102:115-123.
157. Parker JE, Mufti GJ, Rasool F, Mijovic A, Devereux S, Pagliuca A. The role of apoptosis, proliferation, and the Bcl-2-related proteins in the myelodysplastic syndromes and acute myeloid leukemia secondary to MDS. Blood. 2000; 96:3932-3938.
158. Qi H, Xiao L, Lingyun W, Ying T, Yi-Zhi L, Shao-Xu Y, Quan P. Expression of type 1 insulin-like growth factor receptor in marrow nucleated cells in malignant hematological disorders: correlation with apoptosis. Ann Hematol. 2006; 85:95-101.
159. Kim MS, Kim SY, Arunachalam S, Hwang PH, Yi HK, Nam SY, Lee DY. c-myb stimulates cell growth by regulation of insulin-like growth factor (IGF) and IGF-binding protein-3 in K562 leukemia cells. Biochem Biophys Res Commun. 2009; 385:38-43.
160. Schillaci R, Galeano A, Becu-Villalobos D, Spinelli O, Sapia S, Bezares RF. Autocrine/paracrine involvement of insulin-like growth factor-I and its receptor in chronic lymphocytic leukaemia. Br J Haematol. 2005; 130:58-66.
161. Maura F, Mosca L, Fabris S, Cutrona G, Matis S, Lionetti M, Agnelli L, Barbieri M, D’Anca M, Manzoni M, Colombo M, Massucco C, Reverberi D et al. Insulin growth factor 1 receptor expression is associated with NOTCH1 mutation, trisomy 12 and aggressive clinical course in chronic lymphocytic leukemia. PLoS One. 2015; 10:e0118801.
162. Lee PD, Rosenfeld RG, Hintz RL, Smith SD. Characterization of insulin, insulin-like growth factors I and II, and growth hormone receptors on human leukemic lymphoblasts. J Clin Endocrinol Metab. 1986; 62:28-35.
163. Leahy M, Lyons A, Krause D, O’Connor R. Impaired Shc, Ras, and MAPK activation but normal Akt activation in FL5.12 cells expressing an insulin-like growth factor I receptor mutated at tyrosines 1250 and 1251. J Biol Chem. 2004; 279:18306-18313.
164. O’Connor R, Kauffmann-Zeh A, Liu Y, Lehar S, Evan GI, Baserga R, Blattler WA. Identification of domains of the insulin-like growth factor I receptor that are required for protection from apoptosis. Mol Cell Biol. 1997; 17:427-435.
165. Blakesley VA, Koval AP, Stannard BS, Scrimgeour A, LeRoith D. Replacement of tyrosine 1251 in the carboxyl terminus of the insulin-like growth factor-I receptor disrupts the actin cytoskeleton and inhibits proliferation and anchorage-independent growth. J Biol Chem. 1998; 273:18411-18422.
166. Miura M, Surmacz E, Burgaud JL, Baserga R. Different effects on mitogenesis and transformation of a mutation at tyrosine 1251 of the insulin-like growth factor I receptor. J Biol Chem. 1995; 270:22639-22644.
167. Gusscott S, Kuchenbauer F, Humphries RK, Weng AP. Notch-mediated repression of miR-223 contributes to IGF1R regulation in T-ALL. Leuk Res. 2012; 36:905-911.
168. Li XJ, Luo XQ, Han BW, Duan FT, Wei PP, Chen YQ. MicroRNA-100/99a, deregulated in acute lymphoblastic leukaemia, suppress proliferation and promote apoptosis by regulating the FKBP51 and IGF1R/mTOR signalling pathways. Br J Cancer. 2013; 109:2189-2198.
169. Correa PN, Eskinazi D, Axelrad AA. Circulating erythroid progenitors in polycythemia vera are hypersensitive to insulin-like growth factor-1 in vitro: studies in an improved serum-free medium. Blood. 1994;83:99-112.
170. Mirza AM, Correa PN, Axelrad AA. Increased basal and induced tyrosine phosphorylation of the insulin-like growth factor I receptor beta subunit in circulating mononuclear cells of patients with polycythemia vera. Blood. 1995;86:877-882.
171. Mirza AM, Ezzat S, Axelrad AA. Insulin-like growth factor binding protein-1 is elevated I patients with polycythemia vera and stimulates erythroid burst formation. Blood. 1997;89:1862-1869.
172. Michl P, Spoettl G, Engelhardt D, Weber MM. Alterations of the insulin-like growth factor system in patients with polycythemia vera. Mol Cell Endocrinol. 2001; 181:189-197.
173. Staerk J, Kallin A, Demoulin JB, Vainchenker W, Constantinescu SN. JAK1 and Tyk2 activation by the homologous polycythemia vera JAK2 V617F mutation: cross-talk with IGF1 receptor. J Biol Chem. 2005; 280:41893-41899.
174. Osorio FG, Soria-Valles C, Santiago-Fernández O, Bernal T, Mittelbrunn M, Colado E, Rodriguez F, Bonzon-Kulichenko E, Vázquez J, Porta-de-la-Riva M, Cerón J, Fueyo A, Li J et al. Loss of the proteostasis factor AIRAPL causes myeloid transformation by deregulating IGF-1 signaling. Nat Med. 2016; 22:91-96.
175. Weigent DA, Arnold RE. Expression of insulin-like growth factor-1 and insulin-like growth factor-1 receptors in EL4 lymphoma cells overexpressing growth hormone. Cell Immunol. 2005; 234:54-66.
176. Quesada AE, Nguyen ND, Rios A, Brown RE. Morphoproteomics identifies constitutive activation of the mTORC2/Akt and NF-kB pathways and expressions of IGF-1R, Sirt1, COX-2, and FASN in peripheral T-cell lymphomas: pathogenetic implications and therapeutic options. Int J Clin Exp Pathol. 2014; 7:8732-8739.
177. Sabbatini P, Korenchuk S, Rowand JL, Groy A, Liu Q, Leperi D, Atkins C, Dumble M, Yang J, Anderson K, Kruger RG, Gontarek RR, Maksimchuk KR et al. GSK1838705A inhibits the insulin-like growth factor-1 receptor and anaplastic lymphoma kinase and shows antitumor activity in experimental models of human cancers. Mol Cancer Ther. 2009; 8:2811-2820.
178. Vishwamitra D, Curry CV, Alkan S, Song YH, Gallick GE, Kaseb AO, Shi P, Amin HM. The transcription factors Ik-1 and MZF1 downregulate IGF-IR expression in NPM-ALK+ T-cell lymphoma. Mol Cancer. 2015; 14:53.
179. Liang Z, Diepstra A, Xu C, van Imhoff G, Plattel W, Van Den Berg A, Visser L. Insulin-like growth factor 1 receptor is a prognostic factor in classical Hodgkin lymphoma. PLoS One. 2014; 9:e87474.
180. Koh YW, Yoon DH, Suh C, Cha HJ, Huh J. Insulin-like growth factor-1 receptor is associated with better prognosis in classical Hodgkin’s lymphoma: Correlation with MET expression. Int J Exp Pathol. 2015.
181. Stromberg T, Feng X, Delforoush M, Berglund M, Lin Y, Axelson M, Larsson O, Georgii-Hemming P, Lennartsson J, Enblad G. Picropodophyllin inhibits proliferation and survival of diffuse large B-cell lymphoma cells. Med Oncol. 2015; 32:188.
182. Gao J, Chang YS, Jallal B, Viner J. Targeting the insulin-like growth factor axis for the development of novel therapeutics in oncology. Cancer Res. 2012; 72:3-12.
183. Wilky BA, Rudek MA, Ahmed S, Laheru DA, Cosgrove D, Donehower RC, Nelkin B, Ball D, Doyle LA, Chen H, Ye X, Bigley G, Womack C et al. A phase I trial of vertical inhibition of IGF signalling using cixutumumab, an anti-IGF-1R antibody, and selumetinib, an MEK 1/2 inhibitor, in advanced solid tumours. Br J Cancer. 2015; 112:24-31.
184. Ma CX, Suman VJ, Goetz M, Haluska P, Moynihan T, Nanda R, Olopade O, Pluard T, Guo Z, Chen HX, Erlichman C, Ellis MJ, Fleming GF. A phase I trial of the IGF-1R antibody Cixutumumab in combination with temsirolimus in patients with metastatic breast cancer. Breast Cancer Res Treat. 2013; 139:145-153.
185. Reidy DL, Vakiani E, Fakih MG, Saif MW, Hecht JR, Goodman-Davis N, Hollywood E, Shia J, Schwartz J, Chandrawansa K, Dontabhaktuni A, Youssoufian H, Solit DB et al. Randomized, phase II study of the insulin-like growth factor-1 receptor inhibitor IMC-A12, with or without cetuximab, in patients with cetuximab- or panitumumab-refractory metastatic colorectal cancer. J Clin Oncol. 2010; 28:4240-4246.
186. Reidy-Lagunes DL, Vakiani E, Segal MF, Hollywood EM, Tang LH, Solit DB, Pietanza MC, Capanu M, Saltz LB. A phase 2 study of the insulin-like growth factor-1 receptor inhibitor MK-0646 in patients with metastatic, well-differentiated neuroendocrine tumors. Cancer. 2012; 118:4795-4800.
187. Molife LR, Fong PC, Paccagnella L, Reid AH, Shaw HM, Vidal L, Arkenau HT, Karavasilis V, Yap TA, Olmos D, Spicer J, Postel-Vinay S, Yin D et al. The insulin-like growth factor-I receptor inhibitor figitumumab (CP-751,871) in combination with docetaxel in patients with advanced solid tumours: results of a phase Ib dose-escalation, open-label study. Br J Cancer. 2010; 103:332-339.
188. Descamps G, Gomez-Bougie P, Venot C, Moreau P, Bataille R, Amiot M. A humanised anti-IGF-1R monoclonal antibody (AVE1642) enhances Bortezomib-induced apoptosis in myeloma cells lacking CD45. Br J Cancer. 2009; 100:366-369.
189. Sachdev D, Zhang X, Matise I, Gaillard-Kelly M, Yee D. The type I insulin-like growth factor receptor regulates cancer metastasis independently of primary tumor growth by promoting invasion and survival. Oncogene. 2010; 29:251-262.
190. Geoerger B, Brasme JF, Daudigeos-Dubus E, Opolon P, Venot C, Debussche L, Vrignaud P, Vassal G. Anti-insulin-like growth factor 1 receptor antibody EM164 (murine AVE1642) exhibits anti-tumour activity alone and in combination with temozolomide against neuroblastoma. Eur J Cancer. 2010; 46:3251-3262.
191. Spiliotaki M, Markomanolaki H, Mela M, Mavroudis D, Georgoulias V, Agelaki S. Targeting the insulin-like growth factor I receptor inhibits proliferation and VEGF production of non-small cell lung cancer cells and enhances paclitaxel-mediated anti-tumor effect. Lung Cancer. 2011; 73:158-165.
192. Moreau P, Cavallo F, Leleu X, Hulin C, Amiot M, Descamps G, Facon T, Boccadoro M, Mignard D, Harousseau JL. Phase I study of the anti insulin-like growth factor 1 receptor (IGF-1R) monoclonal antibody, AVE1642, as single agent and in combination with bortezomib in patients with relapsed multiple myeloma. Leukemia. 2011; 25:872-874.
193. Macaulay VM, Middleton MR, Protheroe AS, Tolcher A, Dieras V, Sessa C, Bahleda R, Blay JY, LoRusso P, Mery-Mignard D, Soria JC. Phase I study of humanized monoclonal antibody AVE1642 directed against the type 1 insulin-like growth factor receptor (IGF-1R), administered in combination with anticancer therapies to patients with advanced solid tumors. Ann Oncol. 2013; 24:784-791.
194. Soria JC, Massard C, Lazar V, Ozoux ML, Mery-Mignard D, Deslandes A, Tolcher AW. A dose finding, safety and pharmacokinetic study of AVE1642, an anti-insulin-like growth factor-1 receptor (IGF-1R/CD221) monoclonal antibody, administered as a single agent and in combination with docetaxel in patients with advanced solid tumours. Eur J Cancer. 2013; 49:1799-1807.
195. Barnes CJ, Oshiro K, Rayala SK, El-Naggar AK, Kumar R. Insulin-like growth factor receptor as a therapeutic target in head and neck cancer. Clin Cancer Res. 2007; 13:4291-4299.
196. Attias-Geva Z, Bentov I, Ludwig DL, Fishman A, Bruchim I, Werner H. Insulin-like growth factor-I receptor (IGF-IR) targeting with monoclonal antibody cixutumumab (IMC-A12) inhibits IGF-I action in endometrial cancer cells. Eur J Cancer. 2011; 47:1717-1726.
197. Riesterer O, Yang Q, Raju U, Torres M, Molkentine D, Patel N, Valdecanas D, Milas L, Ang KK. Combination of anti-IGF-1R antibody A12 and ionizing radiation in upper respiratory tract cancers. Int J Radiat Oncol Biol Phys. 2011; 79:1179-1187.
198. Zamykal M, Martens T, Matschke J, Gunther HS, Kathagen A, Schulte A, Peters R, Westphal M, Lamszus K. Inhibition of intracerebral glioblastoma growth by targeting the insulin-like growth factor 1 receptor involves different context-dependent mechanisms. Neuro Oncol. 2015; 17:1076-1085.
199. Abou-Alfa GK, Capanu M, O’Reilly EM, Ma J, Chou JF, Gansukh B, Shia J, Kalin M, Katz S, Abad L, Reidy-Lagunes DL, Kelsen DP, Chen HX et al. A phase II study of cixutumumab (IMC-A12, NSC742460) in advanced hepatocellular carcinoma. J Hepatol. 2014; 60:319-324.
200. Malempati S, Weigel B, Ingle AM, Ahern CH, Carroll JM, Roberts CT, Reid JM, Schmechel S, Voss SD, Cho SY, Chen HX, Krailo MD, Adamson PC et al. Phase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol. 2012; 30:256-262.
201. Naing A, Lorusso P, Fu S, Hong D, Chen HX, Doyle LA, Phan AT, Habra MA, Kurzrock R. Insulin growth factor receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with metastatic adrenocortical carcinoma. Br J Cancer. 2013; 108:826-830.
202. Philip PA, Goldman B, Ramanathan RK, Lenz HJ, Lowy AM, Whitehead RP, Wakatsuki T, Iqbal S, Gaur R, Benedetti JK, Blanke CD. Dual blockade of epidermal growth factor receptor and insulin-like growth factor receptor-1 signaling in metastatic pancreatic cancer: phase Ib and randomized phase II trial of gemcitabine, erlotinib, and cixutumumab versus gemcitabine plus erlotinib (SWOG S0727). Cancer. 2014; 120:2980-2985.
203. Vasilcanu D, Girnita A, Girnita L, Vasilcanu R, Axelson M, Larsson O. The cyclolignan PPP induces activation loop-specific inhibition of tyrosine phosphorylation of the insulin-like growth factor-1 receptor. Link to the phosphatidyl inositol-3 kinase/Akt apoptotic pathway. Oncogene. 2004; 23:7854-7862.
204. Girnita A, All-Ericsson C, Economou MA, Astrom K, Axelson M, Seregard S, Larsson O, Girnita L. The insulin-like growth factor-I receptor inhibitor picropodophyllin causes tumor regression and attenuates mechanisms involved in invasion of uveal melanoma cells. Clin Cancer Res. 2006; 12:1383-1391.
205. Duan Z, Choy E, Harmon D, Yang C, Ryu K, Schwab J, Mankin H, Hornicek FJ. Insulin-like growth factor-I receptor tyrosine kinase inhibitor cyclolignan picropodophyllin inhibits proliferation and induces apoptosis in multidrug resistant osteosarcoma cell lines. Mol Cancer Ther. 2009; 8:2122-2130.
206. Klinakis A, Szabolcs M, Chen G, Xuan S, Hibshoosh H, Efstratiadis A. Igf1r as a therapeutic target in a mouse model of basal-like breast cancer. Proc Natl Acad Sci U S A. 2009; 106:2359-2364.
207. Ohshima-Hosoyama S, Hosoyama T, Nelon LD, Keller C. IGF-1 receptor inhibition by picropodophyllin in medulloblastoma. Biochem Biophys Res Commun. 2010; 399:727-732.
208. Tomizawa M, Shinozaki F, Sugiyama T, Yamamoto S, Sueishi M, Yoshida T. Insulin-like growth factor-I receptor in proliferation and motility of pancreatic cancer. World J Gastroenterol. 2010; 16:1854-1858.
209. Yin S, Girnita A, Stromberg T, Khan Z, Andersson S, Zheng H, Ericsson C, Axelson M, Nister M, Larsson O, Ekstrom TJ, Girnita L. Targeting the insulin-like growth factor-1 receptor by picropodophyllin as a treatment option for glioblastoma. Neuro Oncol. 2010; 12:19-27.
210. Feng X, Aleem E, Lin Y, Axelson M, Larsson O, Stromberg T. Multiple antitumor effects of picropodophyllin in colon carcinoma cell lines: clinical implications. Int J Oncol. 2012; 40:1251-1258.
211. Lu X, Wang L, Mei J, Wang X, Zhu X, Zhang Q, Lv J. Picropodophyllin inhibits epithelial ovarian cancer cells in vitro and in vivo. Biochem Biophys Res Commun. 2013; 435:385-390.
212. Qi L, Toyoda H, Shankar V, Sakurai N, Amano K, Kihira K, Iwasa T, Deguchi T, Hori H, Azuma E, Gabazza EC, Komada Y. Heterogeneity of neuroblastoma cell lines in insulin-like growth factor 1 receptor/Akt pathway-mediated cell proliferative responses. Cancer Sci. 2013; 104:1162-1171.
213. Bieghs L, Lub S, Fostier K, Maes K, Van Valckenborgh E, Menu E, Johnsen HE, Overgaard MT, Larsson O, Axelson M, Nyegaard M, Schots R, Jernberg-Wiklund H et al. The IGF-1 receptor inhibitor picropodophyllin potentiates the anti-myeloma activity of a BH3-mimetic. Oncotarget. 2014; 5:11193-11208. doi:10.18632/oncotarget.1933.
214. Zhang Q, Pan J, Lubet RA, Wang Y, You M. Targeting the insulin-like growth factor-1 receptor by picropodophyllin for lung cancer chemoprevention. Mol Carcinog. 2015; 54 Suppl 1:E129-137.
215. Chang TS, Wu YC, Chi CC, Su WC, Chang PJ, Lee KF, Tung TH, Wang J, Liu JJ, Tung SY, Kuo LM, Ho HN, Ling TY et al. Activation of IL6/IGFIR confers poor prognosis of HBV-related hepatocellular carcinoma through induction of OCT4/NANOG expression. Clin Cancer Res. 2015; 21:201-210.
216. Wu YT, Wang BJ, Miao SW, Gao JJ. Picropodophyllin inhibits the growth of Ewing’s sarcoma cells through the insulin-like growth factor-1 receptor/Akt signaling pathway. Mol Med Rep. 2015; 12:7045-7050.
217. Holgersson G, Bergström S, Harmenberg J, Ringbom M, Klockare M, Jerling M, Ekman S, Lundström KL, Koyi H, Brandén E, Larsson O, Bergqvist M. A phase I pilot study of the insulin-like growth factor 1 receptor pathway modulator AXL1717 in combination with gemcitabine HCl and carboplatin in previously untreated, locally advanced, or metastatic non-small cell lung cancer. Med Oncol. 2015; 32:129.
218. Garcia-Echeverria C, Pearson MA, Marti A, Meyer T, Mestan J, Zimmermann J, Gao J, Brueggen J, Capraro HG, Cozens R, Evans DB, Fabbro D, Furet P et al. In vivo antitumor activity of NVP-AEW541-A novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell. 2004; 5:231-239.
219. Scotlandi K, Manara MC, Nicoletti G, Lollini PL, Lukas S, Benini S, Croci S, Perdichizzi S, Zambelli D, Serra M, Garcia-Echeverria C, Hofmann F, Picci P. Antitumor activity of the insulin-like growth factor-I receptor kinase inhibitor NVP-AEW541 in musculoskeletal tumors. Cancer Res. 2005; 65:3868-3876.
220. Gotlieb WH, Bruchim I, Gu J, Shi Y, Camirand A, Blouin MJ, Zhao Y, Pollak MN. Insulin-like growth factor receptor I targeting in epithelial ovarian cancer. Gynecol Oncol. 2006; 100:389-396.
221. Hopfner M, Huether A, Sutter AP, Baradari V, Schuppan D, Scherubl H. Blockade of IGF-1 receptor tyrosine kinase has antineoplastic effects in hepatocellular carcinoma cells. Biochem Pharmacol. 2006; 71:1435-1448.
222. Hopfner M, Sutter AP, Huether A, Baradari V, Scherubl H. Tyrosine kinase of insulin-like growth factor receptor as target for novel treatment and prevention strategies of colorectal cancer. World J Gastroenterol. 2006; 12:5635-5643.
223. Tanno B, Mancini C, Vitali R, Mancuso M, McDowell HP, Dominici C, Raschella G. Down-regulation of insulin-like growth factor I receptor activity by NVP-AEW541 has an antitumor effect on neuroblastoma cells in vitro and in vivo. Clin Cancer Res. 2006; 12:6772-6780.
224. Manara MC, Landuzzi L, Nanni P, Nicoletti G, Zambelli D, Lollini PL, Nanni C, Hofmann F, Garcia-Echeverria C, Picci P, Scotlandi K. Preclinical in vivo study of new insulin-like growth factor-I receptor—specific inhibitor in Ewing’s sarcoma. Clin Cancer Res. 2007; 13:1322-1330.
225. Meyer GE, Chesler L, Liu D, Gable K, Maddux BA, Goldenberg DD, Youngren JF, Goldfine ID, Weiss WA, Matthay KK, Rosenthal SM. Nordihydroguaiaretic acid inhibits insulin-like growth factor signaling, growth, and survival in human neuroblastoma cells. J Cell Biochem. 2007; 102:1529-1541.
226. Slomiany MG, Black LA, Kibbey MM, Tingler MA, Day TA, Rosenzweig SA. Insulin-like growth factor-1 receptor and ligand targeting in head and neck squamous cell carcinoma. Cancer Lett. 2007; 248:269-279.
227. Urbanska K, Trojanek J, Del Valle L, Eldeen MB, Hofmann F, Garcia-Echeverria C, Khalili K, Reiss K. Inhibition of IGF-I receptor in anchorage-independence attenuates GSK-3beta constitutive phosphorylation and compromises growth and survival of medulloblastoma cell lines. Oncogene. 2007; 26:2308-2317.
228. Cunningham MP, Thomas H, Marks C, Green M, Fan Z, Modjtahedi H. Co-targeting the EGFR and IGF-IR with anti-EGFR monoclonal antibody ICR62 and the IGF-IR tyrosine kinase inhibitor NVP-AEW541 in colorectal cancer cells. Int J Oncol. 2008; 33:1107-1113.
229. Friedrichs N, Kuchler J, Endl E, Koch A, Czerwitzki J, Wurst P, Metzger D, Schulte JH, Holst MI, Heukamp LC, Larsson O, Tanaka S, Kawai A et al. Insulin-like growth factor-1 receptor acts as a growth regulator in synovial sarcoma. J Pathol. 2008; 216:428-439.
230. Maiso P, Ocio EM, Garayoa M, Montero JC, Hofmann F, Garcia-Echeverria C, Zimmermann J, Pandiella A, San Miguel JF. The insulin-like growth factor-I receptor inhibitor NVP-AEW541 provokes cell cycle arrest and apoptosis in multiple myeloma cells. Br J Haematol. 2008; 141:470-482.
231. Moser C, Schachtschneider P, Lang SA, Gaumann A, Mori A, Zimmermann J, Schlitt HJ, Geissler EK, Stoeltzing O. Inhibition of insulin-like growth factor-I receptor (IGF-IR) using NVP-AEW541, a small molecule kinase inhibitor, reduces orthotopic pancreatic cancer growth and angiogenesis. Eur J Cancer. 2008; 44:1577-1586.
232. Tarn C, Rink L, Merkel E, Flieder D, Pathak H, Koumbi D, Testa JR, Eisenberg B, von Mehren M, Godwin AK. Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proc Natl Acad Sci U S A. 2008; 105:8387-8392.
233. Adachi Y, Li R, Yamamoto H, Min Y, Piao W, Wang Y, Imsumran A, Li H, Arimura Y, Lee CT, Imai K, Carbone DP, Shinomura Y. Insulin-like growth factor-I receptor blockade reduces the invasiveness of gastrointestinal cancers via blocking production of matrilysin. Carcinogenesis. 2009; 30:1305-1313.
234. Baumann P, Hagemeier H, Mandl-Weber S, Franke D, Schmidmaier R. Myeloma cell growth inhibition is augmented by synchronous inhibition of the insulin-like growth factor-1 receptor by NVP-AEW541 and inhibition of mammalian target of rapamycin by Rad001. Anticancer Drugs. 2009; 20:259-266.
235. Mukohara T, Shimada H, Ogasawara N, Wanikawa R, Shimomura M, Nakatsura T, Ishii G, Park JO, Janne PA, Saijo N, Minami H. Sensitivity of breast cancer cell lines to the novel insulin-like growth factor-1 receptor (IGF-1R) inhibitor NVP-AEW541 is dependent on the level of IRS-1 expression. Cancer Lett. 2009; 282:14-24.
236. Hagerstrand D, Lindh MB, Pena C, Garcia-Echeverria C, Nister M, Hofmann F, Ostman A. PI3K/PTEN/Akt pathway status affects the sensitivity of high-grade glioma cell cultures to the insulin-like growth factor-1 receptor inhibitor NVP-AEW541. Neuro Oncol. 2010; 12:967-975.
237. Bielen A, Perryman L, Box GM, Valenti M, de Haven Brandon A, Martins V, Jury A, Popov S, Gowan S, Jeay S, Raynaud FI, Hofmann F, Hargrave D et al. Enhanced efficacy of IGF1R inhibition in pediatric glioblastoma by combinatorial targeting of PDGFRalpha/beta. Mol Cancer Ther. 2011; 10:1407-1418.
238. Bao XH, Takaoka M, Hao HF, Wang ZG, Fukazawa T, Yamatsuji T, Sakurama K, Sun DS, Nagasaka T, Fujiwara T, Naomoto Y. Esophageal cancer exhibits resistance to a novel IGF-1R inhibitor NVP-AEW541 with maintained RAS-MAPK activity. Anticancer Res. 2012; 32:2827-2834.
239. Bruchim I, Sarfstein R, Reiss A, Flescher E, Werner H. IGF1R tyrosine kinase inhibitor enhances the cytotoxic effect of methyl jasmonate in endometrial cancer. Cancer Lett. 2014; 352:214-219.
240. Rubinfeld H, Kammer A, Cohen O, Gorshtein A, Cohen ZR, Hadani M, Werner H, Shimon I. IGF1 induces cell proliferation in human pituitary tumors - functional blockade of IGF1 receptor as a novel therapeutic approach in non-functioning tumors. Mol Cell Endocrinol. 2014; 390:93-101.
241. Cao L, Yu Y, Darko I, Currier D, Mayeenuddin LH, Wan X, Khanna C, Helman LJ. Addiction to elevated insulin-like growth factor I receptor and initial modulation of the AKT pathway define the responsiveness of rhabdomyosarcoma to the targeting antibody. Cancer Res. 2008; 68: 8039-8048.
242. Zha J, O’Brien C, Savage H, Huw LY, Zhong F, Berry L, Lewis Phillips GD, Luis E, Cavet G, Hu X, Almer LC, Lackner MR. Molecular predictors of response to a humanized anti-insulin-like growth factor-I receptor monoclonal antibody in breast cancer and colorectal cancer. Mol Cancer Ther. 2009; 8:2110-2121.
243. Huang F, Hurlburt W, Greer A, Reeves KA, Hillerman S, Chang H, Fargnoli J, Finckenstein FG, Gottardis MM, Carboni JM. Differential mechanisms of acquired resistance to insulin-like growth factor-1 receptor antibody therapy or to a small-molecule inhibitor, BMS-754807, in a human rhabdomyosarcoma model. Cancer Res. 2010; 70: 7221-7231.
244. Garofalo C, Mancarella C, Grilli A, Manara MC, Astolfi A, Marino MT, Conte A, Sigismund S, Care A, Belfiore A, Picci P, Scotlandi K. Identification of common and distinctive mechanisms of resistance to different anti-IGF-IR agents in Ewing’s sarcoma. Mol Endocrinol. 2012; 26: 1603-1616.
245. Kalra N, Zhang J, Yu Y, Ho M, Merino M, Cao L, Hassan R. Efficacy of anti-insulin-like growth factor I receptor monoclonal antibody cixutumumab in mesothelioma is highly correlated with insulin growth factor-I receptor sites/cell. Int J Cancer. 2012; 131:2143-2152.
246. Sacco A, Morcavallo A, Pandini G, Vigneri R, Belfiore A. Differential signaling activation by insulin and insulin-like growth factors I and II upon binding to insulin receptor isoform A. Endocrinology. 2009; 150:3594-3602.
247. Ulanet DB, Ludwig DL, Kahn CR, Hanahan D. Insulin receptor functionally enhances multistage tumor progression and conveys intrinsic resistance to IGF-1R targeted therapy. Proc Natl Acad Sci USA. 2010; 24: 10791-10798.
248. Forest A, Amatulli M, Ludwig DL, Damoci CB, Wang Y, Burns CA, Donoho GP, Zanella N, Fiebig HH, Prewett MC, Surguladze D, DeLigio JT, Houghton PJ, et al. Intrinsic resistance to cixutumumab is conferred by distinct isoforms of the insulin receptor. Mol Cancer Ther. 2015; 13: 1615-1626.
249. Haluska P, Carboni JM, TenEyck C, Attar RM, Hou X, Yu C, Sagar M, Wong TW, Gottardis MM, Erlichman C. HER receptor signaling confers resistance to the insulin-like growth factor-I receptor inhibitor, BMS-536924. Mol Cancer Ther. 2008; 7:2589-2598.
250. Desbois-Mouthon C, Baron A, Blivet-Van Eggelpoel MJ, Fartoux L, Venot C, Bladt F, Housset C, Rosmorduc O. Insulin-like growth factor-1 receptor inhibition induces a resistance mechanism via the epidermal growth factor receptor/HER3/AKT signaling pathway: rational basis for cotargeting insulin-like growth factor-1 receptor and epidermal growth factor receptor in hepatocellular carcinoma. Clin Cancer Res. 2009; 15:5445-5456.
251. Huang F, Greer A, Hurlburt W, Han X, Hafezi R, Wittenberg GM, Reeves K, Chen J, Robinson D, Li A, Lee FY, Gottardis MM, Clark E, et al. The mechanisms of differential sensitivity to an insulin-like growth factor-1 receptor inhibitor (BMS-536924) and rationale for combining with EGFR/HER2 inhibitors. Cancer Res. 2009; 69: 161-170.
252. Shin DH, Min HY, El-Naggar AK, Lippman SM, Glisson B, Lee HY. Akt/mTOR counteract the antitumor activities of cixutumumab, an anti-insulin-like growth factor I receptor monoclonal antibody. Mol Cancer Ther. 2011; 10:2437-2448.
253. Zinn RL, Gardner EE, Marchionni L, Murphy SC, Dobromilskaya I, Hann CL, Rudin CM. ERK phosphorylation is predictive of resistance to IGF-1R inhibition in small cell lung cancer. Mol Cancer Ther. 2013; 12:1131-1139.
254. Wan X, Yeung C, Heske C, Mendoza A, Helman LJ. IGF-1R inhibition activities a YES/SFK bypass resistance pathway: rational basis for co-targeting IGF-1R and Yes/SFK kinase in rhabdomyosarcoma. Neoplasia. 2015; 4:358-366.
255. Shin DH, Lee HJ, Min HY, Choi SP, Lee MS, Lee JW, Johnson FM, Mehta K, Lippman SM, Glisson BS, Lee HY. Combating resistance to anti-IGFR antibody by targeting the integrin beta3-Src pathway. J Natl Cancer Inst. 2013; 105:1558-1570.
256. Beech DJ, Perer E, Helms J, Gratzer A, Deng N. Insulin-like growth factor-I receptor activation blocks doxorubicin cytotoxicity. Oncol Rep. 2003; 10:181-184.
257. Warshamana-Greene GS, Litz J, Buchdunger E, Garcia-Echeverria C, Hofmann F, Krystal GW. The insulin-like growth factor-I receptor kinase inhibitor, NVP-ADW742, sensitizes small cell lung cancer cell lines to the effects of chemotherapy. Clin Cancer Res. 2005; 11:1563-1571.
258. Oh SH, Jin Q, Kim ES, Khuri FR, Lee HY. Insulin-like growth factor-I receptor signaling pathway induces resistance to the apoptotic activities of SCH66336 (lonafarnib) through Akt/mammalian target of rapamycin-mediated increases in survivin expression. Clin Cancer Res. 2008; 14:1581-1589.
259. Zhou H, Rao J, Lin J, Yin B, Sheng H, Lin F, Zhang N, Yang L. The insulin-like growth factor-I receptor kinase inhibitor NVP-ADW742 sensitizes medulloblastoma to the effects of chemotherapy. Oncol Rep. 2011; 25:1565-1571.
260. Koti M, Gooding RJ, Nuin P, Haslehurst A, Crane C, Weberpals J, Childs T, Bryson P, Dharsee M, Evans K, Feilotter HE, Park PC, Squire JA. Identification of the IGF1/PI3K/NF kappaB/ERK gene signalling networks associated with chemotherapy resistance and treatment response in high-grade serous epithelial ovarian cancer. BMC Cancer. 2013; 13:549.
261. Nakamura S, Watanabe H, Miura M, Sasaki T. Effect of the insulin-like growth factor I receptor on ionizing radiation-induced cell death in mouse embryo fibroblasts. Exp Cell Res. 1997; 235:287-294.
262. Turner BC, Haffty BG, Narayanan L, Yuan J, Havre PA, Gumbs AA, Kaplan L, Burgaud L, Carter D, Baserga R, Glazer PM. Insulin-like growth factor-I receptor overexpression mediates cellular radioresistance and local breast cancer recurrence after lumpectomy and radiation. Cancer Res. 1997; 57:3079-3083.
263. Macaulay VM, Salisbury AJ, Bohula EA, Playford MP, Smorodinsky NI, Shiloh Y. Downregulation of the type 1 insulin-like growth factor receptor in mouse melanoma cells is associated with enhanced radiosensitivity and impaired activation of Atm kinase. Oncogene. 2001; 20:4029-4040.
264. Jameson MJ, Beckler AD, Taniguchi LE, Allak A, Vanwagner LB, Lee NG, Thomsen WC, Hubbard MA, Thomas CY. Activation of the inulin-like growth factor-1 receptor induces resistance to epidermal growth factor receptor antagonism in head and neck squamous cell carcinoma. Mol Cancer Ther. 2001; 10:2124-2134.
265. Lu Y, Zi X, Zhao Y, Mascarenhas D, Pollak M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). J Natl Cancer Inst. 2001; 93:1852-1857.
266. Jones HE, Goddard L, Gee JM, Hiscox S, Rubini M, Barrow D, Knowlden JM, Williams S, Wakeling AE. Insulin-like growth factor-I receptor signalling and acquired resistance to gefitinib (ZD1839; Iressa) in human breast and prostate cancer cells. Endocr Relat Cancer. 2004; 11:793-814.
267. Morgillo F, Kim WY, Kim ES, Ciardiello F, Hong WK, Lee HY. Implication of the insulin-like growth factor-IR pathway in the resistance of nom-small lung cancer cells to treatment with gefitinib. Clin Cancer Res. 2007; 13:2795-2803.
268. Yang L, Li J, Ran L, Pan F, Zhao X, Ding Z, Chen Y, Peng Q, Liang H. Phosphorylated insulin-like growth factor 1 receptor is implicated in resistance to the cytostatic effect of gefitinib in colorectal cancer cells. J Gastrointest Surg. 2011; 15:942-957.
269. Suda K, Mizuuchi H, Sato K, Takemoto T, Iwasaki T, Mitsudomi T. The insulin-like growth factor 1 receptor causes acquired resistance to erlotinib in lung cancer cells with the wild-type epidermal growth factor receptor. Int J Cancer. 2014; 135:1002-1006.
270. Murakami A, Takahashi F, Nurwidya F, Kobayashi I, Minakata K, Hashimoto M, Nara T, Kato M, Tajima K, Shimada N, Iwakami S, Moriyama M, Koizumi F et al. Hypoxia increases gefitinib-resistant lung cancer stem cells through the activation of insulin-like growth factor 1 receptor. PLoS One. 2014; 9:e86459.
271. Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005; 65:11118-11128.
272. Morgillo F, Woo JK, Kim ES, Hong WK, Lee HY. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin counteracts the antitumor action of erlotinib. Cancer Res. 2006; 66:10100-10111.
273. Nickerson T, Chang F, Lorimer D, Smeekens SP, Sawyers CL, Pollak M. In vivo progression of LAPC-9 and LNCaP prostate cancer models to androgen independence is associated with increased expression of insulin-like growth factor ( (IGF-I) and IGF-IR receptor (IGF-IR). Cancer Res. 2001; 61:6276-6280.
274. Krueckl SL, Sikes RA, Edlund NM, Bell RH, Hurtado-Coll A, Fazli L, Gleave ME, Cox ME. Increased insulin-like growth factor I receptor expression and signaling are components of androgen-independent progression in a lineage-derived prostate cancer progression model. Cancer Res. 2004; 64:8620-8629.
275. Zhang Y, Moerkens M, Ramaiahgari S, de Bont H, Price L, Meerman J, van de Water B. Elevated insulin-like growth factor 1 receptor signaling induces antiestrogen resistance through the MAPK/ERK and PI3K/Akt signaling routes. Breast Cancer Res. 2011; 13:R52.
276. Fox EM, Kuba MG, Miller TW, Davies BR, Arteaga CL. Autocrine IGF-I/insulin receptor axis compensates for inhibition of AKT in ER-positive breast cancer cells with resistance to estrogen deprivation. Breast Cancer Res. 2013; 15:R55.
277. Li L, Wang Y, Peng T, Zhang K, Lin C, Han R, Lu C, He Y. Metformin restores crizotinib sensitivity in crizotinib-resistant human lung cancer cells through inhibition of IGF1-R signaling pathway. Oncotarget. 2016; 7:34442-52. doi: 10.18632/oncotarget.9120.
278. O’Flanagan CH, O’Shea S, Lyons A, Fogarty FM, McCabe N, Kennedy RD, O’Conner R. IGF-1R inhibition sensitizes breast cancer cells to ATM-related kinase (ATR) inhibitor and cisplatin. Oncotarget. 2016; 7:56826-56841. doi: 10.18632/oncotarget.10862.
279. Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, Holland EC, Sutton JC, Joyce JA. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science. 2016; 352:aad3018. 10.1126/science.aad3018.
280. Sanabria-Figueroa E, Donnelly SM, Foy KC, Buss MC, Castellino RC, Paplomata E, Taliaferro-Smith L, Kaumaya PT, Nahta R. Insulin-like growth factor-1 receptor signaling increases the invasive potential of human epidermal growth factor receptor 2-overexpressing breast cancer cells via Src-focal adhesion kinase and forkhead box protein M1. Mol Pharmacol. 2015; 87:150-161.
281. Chakraborty AK, Zerillo C, DiGiovanna MP. In vitro and in vivo studies of the combination of IGF1R inhibitor figitumumab (CP-751,871) with HER2 inhibitors trastuzumab and neratinib. Breast Cancer Res Treat. 2015; 152:533-544.
282. Macaulay VM, Middleton MR, Eckhardt SG, Rudin CM, Juergens RA, Gedrich R, Gogov S, McCarthy S, Poondru S, Stephens AW, Gadgeel SM. Phase I dose-escalation study of linsitinib (OSI-906) and erlotinib in patients with advanced solid tumors. Clin Cancer Res. 2016; 22:2897-2907.
283. Pappano WN, Jung PM, Meulbroek JA, Wang YC, Hubbard RD, Zhang Q, Grudzien MM, Soni NB, Johnson EF, Sheppard GS, Donawho C, Buchanan FG, Davidsen SK et al. Reversal of oncogene transformation and suppression of tumor growth by the novel IGF1R kinase inhibitor A-928605. BMC Cancer. 2009; 9:314.
284. Jones HE, Gee JM, Barrow D, Tonge D, Holloway B, Nicholson RI. Inhibition of insulin receptor isoform-A signalling restores sensitivity to gefitinib in previously de novo resistant colon cancer cells. Br J Cancer. 2006; 95:172-180.
285. Cosaceanu D, Carapancea M, Alexandru O, Budiu R, Martinsson HS, Starborg M, Vrabete M, Kanter L, Lewensohn R, Dricu A. Comparison of three approaches for inhibiting insulin-like growth factor I receptor and their effects on NSCLC cell lines in vitro. Growth Factors. 2007; 25:1-8.
286. Chakraborty AK, Welsh A, Digiovanna MP. Co-targeting the insulin-like growth factor I receptor enhances growth-inhibitory and pro-apoptotic effects of anti-estrogens in human breast cancer cell lines. Breast Cancer Res Treat. 2010; 120:327-335.
287. Kai K, D’Costa S, Sills RC, Kim Y. Inhibition of the insulin-like growth factor 1 receptor pathway enhances the antitumor effect of cisplatin in human malignant mesothelioma cell lines. Cancer Lett. 2009; 278:49-55.
288. Li P, Veldwijk MR, Zhang Q, Li ZB, Xu WC, Fu S. Co-inhibition of epidermal growth factor receptor and insulin-like growth factor receptor 1 enhances radiosensitivity in human breast cancer cells. BMC Cancer. 2013; 13:297.
289. Luk F, Yu Y, Walsh WR, Yang JL. IGF1R-targeted therapy and its enhancement of doxorubicin chemosensitivity in human osteosarcoma cell lines. Cancer Invest. 2011; 29:521-532.
290. Momose I, Kunimoto S, Osono M, Ikeda D. Inhibitors of insulin-like growth factor-1 receptor tyrosine kinase are preferentially cytotoxic to nutrient-deprived pancreatic cancer cells. Biochem Biophys Res Commun. 2009; 380:171-176.
291. Steinbach JP, Eisenmann C, Klumpp A, Weller M. Co-inhibition of epidermal growth factor receptor and type 1 insulin-like growth factor receptor synergistically sensitizes human malignant glioma cells to CD95L-induced apoptosis. Biochem Biophys Res Commun. 2004; 321:524-530.
292. Wang Y, Yuan JL, Zhang YT, Ma JJ, Xu P, Shi CH, Zhang W, Li YM, Fu Q, Zhu GF, Xue W, Lei YH, Gao JY et al. Inhibition of both EGFR and IGF1R sensitized prostate cancer cells to radiation by synergistic suppression of DNA homologous recombination repair. PLoS One. 2013; 8:e68784.
293. Wen B, Deutsch E, Marangoni E, Frascona V, Maggiorella L, Abdulkarim B, Chavaudra N, Bourhis J. Tyrphostin AG 1024 modulates radiosensitivity in human breast cancer cells. Br J Cancer. 2001; 85:2017-2021.
294. Yao WF, Liu JW, Sheng GL, Huang DS. Blockade of IGF-IR exerts anticancer effects in hepatocellular carcinoma. Mol Med Rep. 2011; 4:719-722.
295. Hou X, Huang F, Carboni JM, Flatten K, Asmann YW, Ten Eyck C, Nakanishi T, Tibodeau JD, Ross DD, Gottardis MM, Erlichman C, Kaufmann SH, Haluska P. Drug efflux by breast cancer resistance protein is a mechanism of resistance to the benzimidazole insulin-like growth factor receptor/insulin receptor inhibitor, BMS-536924. Mol Cancer Ther. 2011; 10:117-125.
296. Lippolis C, Refolo MG, D’Alessandro R, Carella N, Messa C, Cavallini A, Carr BI. Resistance to multikinase inhibitor actions mediated by insulin like growth factor-1. J Exp Clin Cancer Res. 2015; 34:90.
297. Zhou F, Chen X, Fan S, Tai S, Jiang C, Zhang Y, Hao Z, Zhou J, Shi H, Zhang L, Liang C. GSK1838705A, an insulin-like growth factor-1 receptor/insulin receptor inhibitor, induces apoptosis and reduces viability of docetaxel-resistant prostate cancer cells both in vitro and in vivo. Onco Targets Ther. 2015; 8:753-760.
298. Zhou X, Shen F, Ma P, Hui H, Pei S, Chen M, Wang Z, Zhou W, Jin B. GSK1838705A, an IGF-1R inhibitor, inhibits glioma cell proliferation and suppresses tumor growth in vivo. Mol Med Rep. 2015; 12:6541-6546.
299. Sabbatini P, Rowand JL, Groy A, Korenchuk S, Liu Q, Atkins C, Dumble M, Yang J, Anderson K, Wilson BJ, Emmitte KA, Rabindran SK, Kumar R. Antitumor activity of GSK1904529A, a small-molecule inhibitor of the insulin-like growth factor-I receptor tyrosine kinase. Clin Cancer Res. 2009; 15:3058-3067.
300. Ozkan EE. Plasma and tissue insulin-like growth factor-I receptor (IGF-IR) as a prognostic marker for prostate cancer and anti-IGF-IR agents as novel therapeutic strategy for refractory cases: a review. Mol Cell Endocrinol. 2011; 344:1-24.
301. Agullo-Ortuno MT, Diaz-Garcia CV, Agudo-Lopez A, Perez C, Cortijo A, Paz-Ares L, Lopez-Rios F, Pozo F, de Castro J, Cortes-Funes H, Lopez Martin JA. Relevance of insulin-like growth factor 1 receptor gene expression as a prognostic factor in non-small-cell lung cancer. J Cancer Res Clin Oncol. 2015; 141:43-53.
302. Martins AS, Mackintosh C, Martin DH, Campos M, Hernandez T, Ordonez JL, de Alava E. Insulin-like growth factor I receptor pathway inhibition by ADW742, alone or in combination with imatinib, doxorubicin, or vincristine, is a novel therapeutic approach in Ewing tumor. Clin Cancer Res. 2006; 12:3532-3540.
303. Vishwamitra D, Curry CV, Shi P, Alkan S, Amin HM. SUMOylation confers posttranslational stability on NPM-ALK oncogenic protein. Neoplasia. 2015; 17:742-754.