
INTRODUCTION
LRP1B is a member of the LDL receptor family of lipoprotein receptors, which are involved in different functions in the human body including cholesterol metabolism and atherosclerotic lesion formation. LRP1B was originally discovered during the study of lung cancer cell lines [1]. In nearly 50% of non-small cell lung cancer (NSCLC) cell lines, alterations of the LRP1B gene with homozygous deletions of exons or abnormal transcripts missing portions of the LRP1B sequence were observed. Therefore, LRP1B was postulated as a putative tumor suppressor. In subsequent studies, LRP1B was found to be inactivated in multiple malignancies, namely urothelial cancer, hepatobiliary tumors, esophageal carcinoma, cervix carcinoma, glioblastoma, oral squamous cell carcinoma, small B-cell lymphoma, acute lymphoblastic leukemia, gastric cancer, thyroid cancer, melanoma, ovarian cancer, renal cell cancer, and adrenocortical carcinoma [2–15]. Besides allelic loss of heterozygosity and inactive mRNA transcripts, DNA methylation of CpG islands has been described as mechanism leading to decreased LRP1B expression in various tumors [4, 8–11]. Recently, LRP1B was identified as integration site for hepatitis B virus and human papilloma virus presumably with impact on LRP1B expression [16, 17]. Taken together, these observations strongly suggest a role of LRP1B in tumorigenesis and strengthen the original hypothesis of the receptor serving as a tumor suppressor. Recently, we have characterized the expression of LRP1B in normal human tissues, which appears to be mostly restricted to brain, skeletal muscle, thyroid gland and testis. In addition, expression in smooth muscle cells of the arterial wall has been described [18].
LRP1B is one of the largest transmembrane receptors comprising 4599 amino acids encoded by an mRNA of 13800 base pairs. Similar to the homologous LRP1 receptor, LRP1B contains four ligand binding domain regions separated by EGF precursor homology regions, a transmembrane segment and a cytoplasmic tail containing two NPxY motifs [1]. In contrast to the homologous LRP1, LRP1B is not cleaved by furin and therefore migrates as single polypeptide chain with an apparent molecular weight of 600 kD on SDS polyacrylamide gels [19].
To gain insight into the physiological functions of LRP1B, a knockout mouse model has been generated by replacing the transmembrane domain (exon 88) with a neomycin cassette, resulting in the absence of a membrane-inserted receptor. These mice were viable and fertile and did not show any obvious abnormalities, including no increased tumor rate [19]. However, when the Lrp1b gene was inactivated by more proximal deletions, no viable homozygous Lrp1b mutant animals were obtained, strongly suggesting a crucial role for the extracellular domain in normal development [20].
To further characterize the physiological function of the receptor, several attempts have been made to construct a recombinant LRP1B receptor. However, due to the enormous size of the polypeptide chain, only minireceptors comprising a part of the LRP1B sequence (ligand binding domain region IV, transmembrane segment and intracellular tail) and soluble ligand binding ectodomains have been constructed [19, 21]. In the present study we used a PCR-based strategy to construct a recombinant full-length Lrp1b expression vector. This recombinant receptor was then introduced into human cells lacking endogenous LRP1B and cellular proliferation was analyzed. To exclude artifacts caused by overexpression, control experiments using siRNA to silence LRP1B expression were performed.
RESULTS
Amplification and subcloning of N-terminal, middle and C-terminal mLrp1b fragments
Due to the enormous size of the mLrp1b cDNA (13.8 kb, Genebank NM_053011), the coding sequence was divided into three parts (N-terminal (3810 bp), middle (5970 bp) and C-terminal (4020 bp) fragments) and amplified separately from mouse brain cDNA using specific primers (Figure 1). To ensure efficient transcription, a Kozak consensus sequence was included preceding the start codon within the N-terminal fragment. The integrity of the sequences was confirmed by restriction enzyme digestion and complete sequencing. Even with polymerases containing proof reading enzymes, single base substitutions cannot be avoided in these large amplified DNA segments. Therefore, in several instances multiple amplified clones had to be analyzed to obtain sequences without non-conservative base exchanges. We found a particular high rate of base substitutions in a 2213 bp GC-rich region within the middle fragment flanked by two unique restriction sites (BspEI and PmeI). Therefore, this region was amplified separately and then reinserted into the middle fragment.
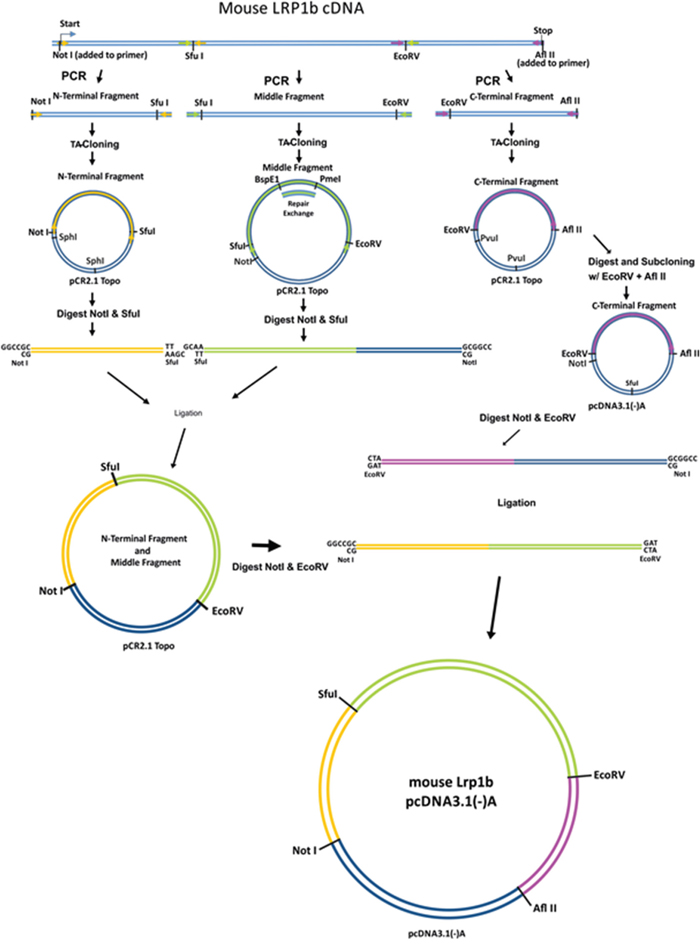
Figure 1: Cloning strategy to construct a full-length Lrp1b receptor. The murine Lrp1b m-RNA was divided into three fragments separated by unique restriction sites and amplified from mouse brain cDNA using specific primers containing the indicated unique restriction sites. After subcloning in a TA vector and repair of a GC rich sequence within the middle fragment, the fragments were joined and cloned into the pcDNA3.1 (-)/myc-His A expression vector. The myc and HIS tag were excised during the cloning process.
Construction and expression of the full-length mLrp1b expression vector
First, the C-terminal fragment was cut out of the TOPO vector using EcoRV(5’) and AflII(3’) followed by degradation of the vector backbone with PvuI, allowing the separation of the insert from the vector of similar size by preparative agarose gel electrophoresis. The C-terminal fragment was then cloned into the mammalian expression vector pcDNA3.1(-)/myc-HisA using the same unique restriction enzyme sites. To avoid a three fragment ligation with large fragments, the N-terminal and middle fragment were subcloned into the pCR2.1 Topo vector. To this end, the vector containing the N-terminal fragment was cut with SfuI and NotI. Also in this case, degradation of the vector backbone with SphI was necessary to allow the separation of the insert from the vector by preparative agarose gel electrophoresis. The N-terminal insert was subsequently ligated into the linearized (NotI and SfuI) middle-insert containing vector.
The last step was to cut the pcDNA3.1 vector containing the C-terminal fragment with EcoRV and NotI, preparing the joined N-terminal and middle fragment insert using the same enzymes, and ligating the two parts to obtain the full-length Lrp1b expression vector (Figure 2A). The integrity of the sequence was confirmed by sequencing of the whole insert using the sequencing primers shown in Supplementary Table S1 thereby confirming the absence of any non-conservative mutations. Due to the usage of the AflII site located between the HIS-tag and the BGH polyadenylation signal, the myc and HIS tag were excised from the vector backbone and the construct therefore codes for the full-length Lrp1b receptor without a C-terminal tag. A comparison of the protein structure of the full length murine Lrp1b protein and the previously established human LRP1B region IV minireceptor [19] is depicted in Figure 2B.
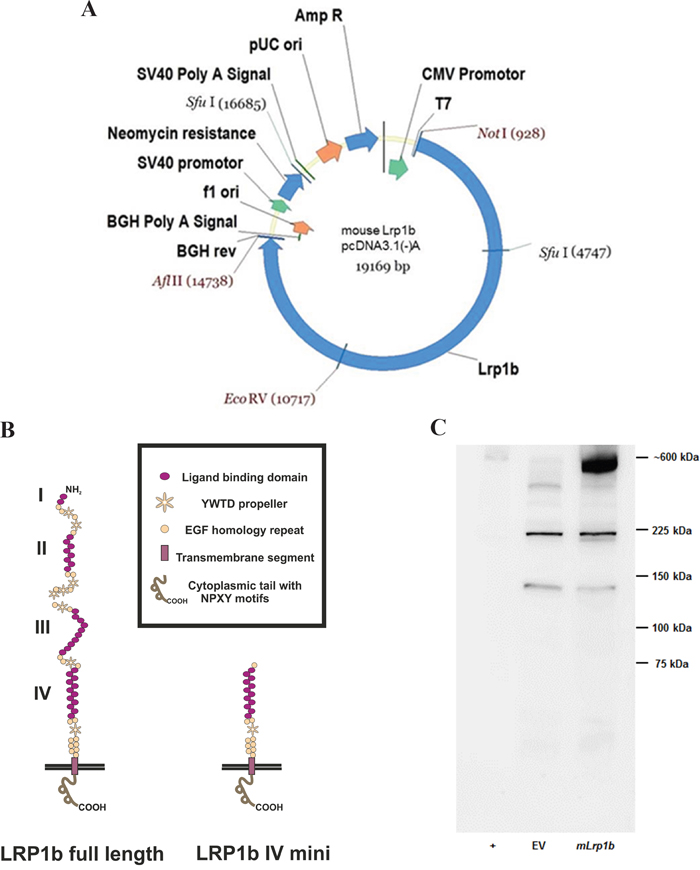
Figure 2: Expression of full-length mLrp1b in HEK 293 cells. A. The pcDNA 3.1 mammalian expression vector containing the full length mLrp1b cDNA is shown with the unique restriction enzyme sites used for cloning. B. Comparison of the protein structure of the full length murine Lrp1b protein and the receptor containing only domain IV (i.e. minireceptor) C. HEK 293 cells were transfected with plasmids containing either full-length mLrp1b or empty vector (EV). Protein lysates were prepared and subjected to Western blotting using a polyclonal antibody directed against a C-terminal epitope. Crude membrane preparations from mouse brain served as positive control (+).
The full-length expression vector and the empty vector as negative control were linearized with NruI and subsequently transfected into HEK 293 cells. Transfected cells were selected by resistance to G418 and tested for effective expression by Western blotting with a C-terminal polyclonal antibody. As shown in Figure 2C, in HEK 293 cells transfected with the mLrp1b construct, a band at approximately 600 kD appeared, corresponding to a band with the same apparent molecular weight in the positive control lane (mouse brain lysate). The lower bands in the mLrp1b-transfected HEK 293 cells are non-specific, since they were also observed in the negative control (empty vector).
Basal LRP1B expression in human non-small cell lung cancer cell lines
To identify cell lines lacking endogenous LRP1B expression, NSCLC cell lines were tested for the presence of LRP1B transcripts using a sensitive quantitative RT-PCR (qPCR) with primers specific for human LRP1B. HEK 293 cells, which have been previously shown to express endogenous LRP1B [22], served as positive control. As shown in Figure 3, two NSCLC cell lines (A427, A549) showed very low LRP1B expression. In another NSCLC cell line (HCC-44), no LRP1B transcripts could be detected with this very sensitive assay. In contrast, another NSCLC cell line (Calu-1) showed relatively high LRP1B expression (about 1/10 of HEK 293 cells).
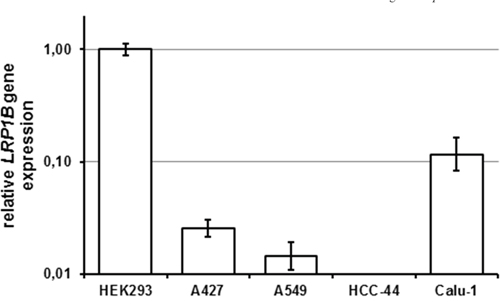
Figure 3: Endogenous LRP1B levels in non-small cell lung cancer cell lines. Endogenous LRP1B mRNA levels in A427, A549, HCC-44 and Calu-1 cells were measured by quantitative PCR. Gene expression levels were normalized using the housekeeping gene RPLP0 and are shown relative to HEK 293 cells. Values are depicted as mean and SEM and are representative for at least three independent experiments run in triplicates.
Expression of full-length Lrp1b inhibits proliferation of non-small cell lung cancer cells
Given their very low endogenous LRP1B expression levels, the NSCLC cell lines A427, A549 and HCC-44 were used to investigate the effect of Lrp1b overexpression on cellular proliferation. Thus, the cell lines were stably transfected with full-length Lrp1b or a human LRP1B minireceptor. The expression of mLrp1b and hLRP1B transcripts in these transfected cells was measured by qPCR using primers specific for the mouse and human sequences, respectively. As shown in Figure 4A, mLrp1b and hLRP1B minireceptor in transfected cells was higher (A427, A549) or at least similar (HCC-44) compared to endogenous LRP1B levels in HEK 293 cells. Efficient expression of full-length mLRP1B protein was proven by immunoblotting using a polyclonal LRP1B-specific antibody (Figure 4B).
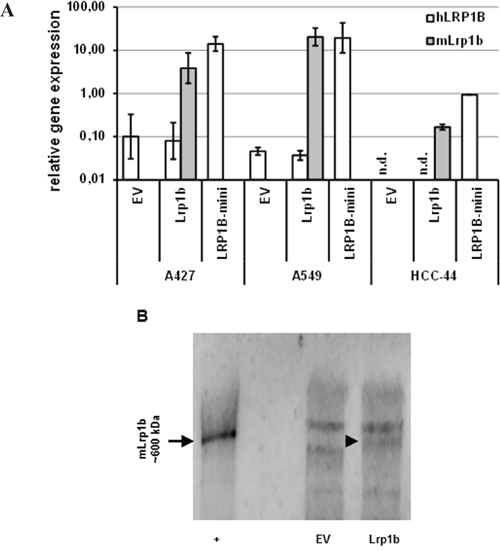
Figure 4: Effective expression of Lrp1b in non-small cell lung cancer cells. A. A427, A549 and HCC-44 cells were stably transfected with plasmids containing either murine full-length Lrp1b or a human LRP1B minireceptor (LRP1B-mini). Empty vector (EV) transfected cells served as control. The level of expression of hLRPB (endogenous and hLRP1B minireceptor) and mLrp1b (full-length mLrp1b) were determined using primers specific for the human (hLRP1B) and mouse (mLrp1b) sequences, respectively. Gene expression levels were normalized using the housekeeping gene RPLP0 and are shown relative to endogenous LRP1B levels of HEK 293 cells. Values are given as mean and SEM and are representative for at least three independent experiments run in triplicates. n.d. not detectable. B. Verification of full-length mLRP1B overexpression at protein levels in A427 cells. Western blot analysis of total cell lysates from Lrp1b transfected cells and empty vector control using a polyclonal rabbit-anti LRP1B antibody (recognizing both the human and mouse sequences). Crude membrane preparations from mouse brain served as positive control (+).
Subsequently, cellular proliferation of stably transfected cells was investigated. In agreement with its proposed tumor suppressor function, overexpression of full-length mLrp1b significantly reduced cellular proliferation of A427, A549 and HCC-44 cells in a BrdU incorporation assay. Stable transfection with the hLRP1B minireceptor resulted in comparable attenuation of proliferation (Figure 5).
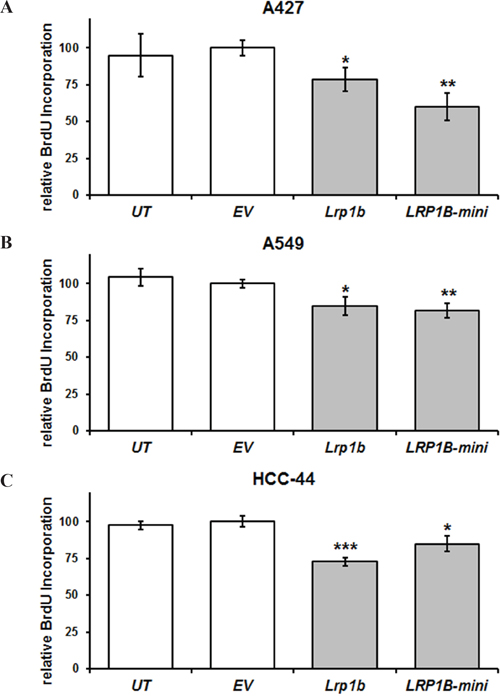
Figure 5: Lrp1b overexpression attenuates cellular proliferation of non-small cell lung cancer cell lines in vitro. Relative quantification of DNA synthesis as determined by bromodeoxyuridine (BrdU) incorporation ELISA is shown for A. A427 B., A549, and C. HCC-44 NSCLC cells stably transfected with plasmids containing either full-length mLrp1b or a hLRP1B minireceptor (LRP1B-mini). Untransfected (UT) and empty vector (EV) transfected cells served as controls. Values are given as mean and SEM and are representative for at least three independent experiments run in triplicates. Significance versus EV is indicated (* P < 0.05, ** P < 0.01, *** P < 0.001).
To investigate if this anti-proliferative effect was specific for LRP1B or could be also mimicked by highly related proteins such as LRP1, a previously established full length LRP1 receptor [23] was employed. However, in contrast to full-length mLrp1b or the hLRP1B minireceptor, LRP1 overexpression did not significantly affect cellular proliferation of A549 (Supplementary Figure S1) and A427 cells (data not shown), respectively.
siRNA-mediated LRP1B knockdown increases cellular proliferation of Calu-1 cells
To verify that the reduced proliferation observed in NSCLC cells expressing mLrp1b was a specific effect of Lrp1b and not due to side effects or overexpression artefacts (e.g. endoplasmatic reticulum stress (unfolded protein response) that is commonly induced by overexpression of highly-glycosylated proteins), siRNA-mediated LRP1B knockdown was applied. To this end, Calu-1 NSCLC cells were selected due to their relatively high endogenous LRP1B levels (Figure 3).
Transfection with LRP1B-specific siRNA resulted in an approximately 65% reduction of LRP1B mRNA levels compared with non-targeting control siRNA (Figure 6A) and effective knockdown was verified at protein levels (Figure 6B). In contrast to the attenuated cellular proliferation of NSCLC cells observed after Lrp1b overexpression, LRP1B knockdown significantly enhanced the proliferation of Calu-1 cells (Figure 6C).
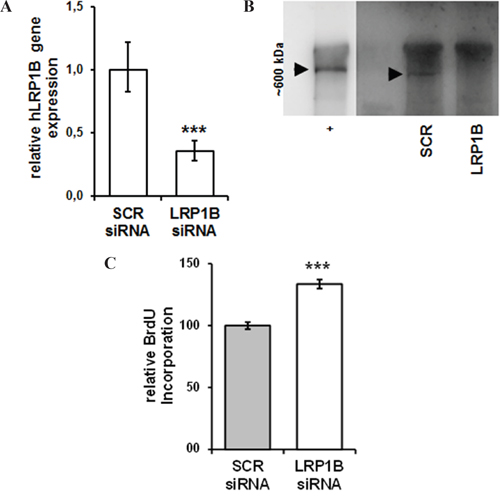
Figure 6: siRNA-mediated knockdown of LRP1B increases Calu-1 proliferation. Calu-1 NSCLC cells were transfected with a pool of LRP1B specific siRNAs or non-target scrambled (SCR) control siRNAs. A. Effective knockdown of LRP1B is shown by quantitative PCR relative to Calu-1 cells treated with scrambled siRNA and normalized using the housekeeping gene RPLP0. B. Western blot analysis of total cell lysates using a polyclonal rabbit-anti LRP1B antibody. Crude membrane preparations from mouse brain served as positive control (+). C. Relative quantification of DNA synthesis as determined by bromodeoxyuridine (BrdU) incorporation ELISA. Values are given as mean and SEM and are representative for at least three independent experiments run in triplicates. Significance versus SCR is indicated (*** P < 0.001).
Expression of full-length Lrp1b increases p21CIP1 levels and Stat3 phosphorylation and reduces Src phosphorylation in A427 cells
To gain first insights into the molecular mechanism underlying the growth attenuation caused by Lrp1b overexpression, the impact of previously identified potential LRP1B ligands on cellular proliferation was investigated. Therefore, A427 cells stably transfected with the full-length mLrp1b or empty vector control were stimulated with various concentrations of very low-density lipoprotein (VLDL) or fibrinogen, putative ligands that have been shown to bind to the minireceptor and/or soluble ectodomains previously [22]. However, while VLDL did not affect proliferation rate and fibrinogen dose-dependently enhanced proliferation of both Lrp1b overexpressing and control cells, Lrp1b overexpression constantly attenuated proliferation rate by 30-40% irrespective of VLDL or fibrinogen supplementation at all concentrations tested (Supplementary Figure S2).
Additionally, several intracellular signaling pathways that potentially control cell proliferation were analyzed in A427 cells by Western blot analysis using (phosphorylation) specific antibodies (Figure 7). mLrp1b overexpression did not significantly affect activation of protein kinase B (AKT) and mitogen-activated protein kinase (MAPK) signaling in response to serum stimulation as determined by levels of phosphorylated AKT and ERK1/2, respectively. However, protein levels of the cyclin-dependent kinase inhibitor 1 (CDKN1A/p21CIP1) were enhanced in cells overexpressing mLrp1b whereas phosphorylation of proto-oncogene tyrosine-protein kinase Src at residue Tyr416 was attenuated. Moreover, phosphorylation of the transcription factor signal transducer and activator of transcription 3 (Stat3) was increased in A427 transfected with the full-length mLrp1b vector compared with empty vector control cells (Figure 7).
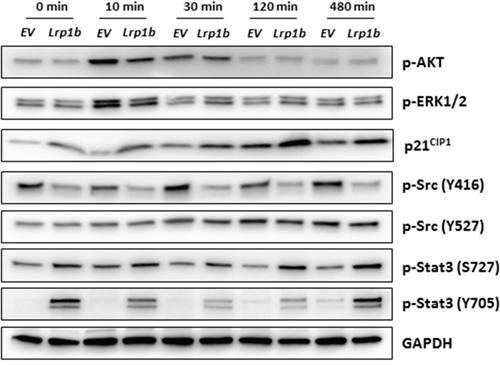
Figure 7: Effect of Lrp1b overexpression on intracellular signalling pathways. A427 cells stably transfected with plasmids containing either murine full-length Lrp1b or empty vector (EV) control were serum-starved overnight and subsequently stimulated with 10% fetal calf serum for different time intervals. Thereafter, total protein lysates were analyzed by Western blotting using the indicated antibodies. GAPDH served as loading control. Images are representative of three independent experiments.
DISCUSSION
Using a PCR-based cloning strategy, we were able to construct a mammalian expression vector encoding the entire murine Lrp1b mRNA (13800 base pairs). The construct was introduced by transfection into human cell lines (HEK 293 and NSCLC cell lines). Using a sensitive qPCR method to detect and discriminate hLRP1B and mLrp1b mRNA sequences, efficient expression of the full-length mLrp1b construct in these cells was proven. Moreover, mLRP1B protein expression was demonstrated by the appearance of an approximately 600 kDa band on Western blots using a specific affinity-purified antibody. As described previously, LRP1B is expressed as a single polypeptide chain and, unlike the homologous LRP1, does not undergo proteolytic cleavage by furin [19]. Consistently, overexpression of mLrp1b resulted in a single band in Western blot analysis without any additional bands that would indicate proteolytic cleavage (Figure 2C). To the best of our knowledge, this represents the largest transmembrane receptor expressed successfully in recombinant form in mammalian cells.
Previous studies that tried to identify extracellular ligands of LRP1B by using LRP1B minireceptors and soluble ectodomain constructs revealed a variety of potential binding partners, including VLDL and fibrinogen, [21, 22, 24]. In addition, several well-known or postulated molecular chaperones were identified by affinity chromatography with these ectodomains, highlighting the important role of chaperones in the biosynthesis and correct folding of this complex receptor [19, 25]. When an LRP1B minireceptor was introduced into mammalian cells, a slower endocytosis rate compared to analogous minireceptor constructs derived from LRP1 has been reported [21, 26]. In addition, a slower internalization rate of uPA/PAI- complexes and a decreased migration rate were described in cells expressing the LRP1B minireceptor compared to cells expressing a homologous LRP1 construct [24]. While minireceptors are very useful tools in identifying ligands or mapping binding sites, they may not necessarily share all biological functions with the full –length receptor molecules [27, 28]. Notably, in the present study the previously identified potential LRP1B ligands VLDL and fibrinogen did not alter the growth-suppressing function of the receptor in A427 cells.
In agreement with the LRP1B inactivation in many malignancies and the thus postulated putative tumor suppressor function [1–15], we identified NSCLC cell lines with very low (A427, A549) or absent (HCC-44) endogenous LRP1B expression. In contrast, in another NSCLC cell line, Calu-1 a relatively high endogenous LRP1B expression was observed.
In NSCLC cells with low/absent endogenous LRP1B levels overexpression of full-length mLrp1b or a LRP1B minireceptor resulted in marked inhibition of cellular proliferation compared to empty vector transfected cells, strongly supporting the proposed tumor suppressor function. Moreover, these findings are in line with previous results demonstrating that a LRP1B minireceptor suppressed anchorage-independent growth of LRP1B-deficient H4 neuroglioma cells [29]. The growth-suppressing function of LRP1B was further corroborated by the fact that siRNA-mediated silencing of endogenous LRP1B in Calu-1 cells significantly increased cellular proliferation. Similarly, knockdown of LRP1B significantly promoted anchorage-independent growth in HEK 293 cells and renal cancer cells 127 [14].
The molecular mechanisms underlying the growth-suppressing function of LRP1B remain elusive. Our findings indicate that this anti-proliferative activity was not affected by addition of the potential receptor ligands VLDL or fibrinogen. Moreover, Lrp1b overexpression appeared not to affect mitogen-stimulated intracellular AKT and MAPK/ERK signaling. However, Lrp1b overexpression increased levels of p21CIP1, a cyclin-dependent kinase inhibitor that blocks cell cycle progression from G1 to S phase [30]. Moreover, phosphorylation of Src at residue Tyr416 was attenuated in Lrp1b expressing A427 cells. Src represents an important proto-oncogene that promotes cellular proliferation. Scr activation has been documented in more than 50% of tumors derived from colon, liver, lung, breast, and pancreas and [31]. Of note, whereas overexpression of Lrp1b appeared to inactivate Src previous findings demonstrated that Src can be activated by LRP1 [32].
Given that constitutive activation of Stat3 is a frequent aberrancy in cancer cells that is supposed to directly contribute to tumorigenesis, the observed activation of Stat3 in A427 cells overexpressing the proposed tumor suppressor Lrp1b appears contradictory. However, newer studies indicated that Stat3 exerts also tumor suppressor effects under specific conditions. As reviewed recently, Stat3 can function either as an oncoprotein or a tumor suppressor in the same cell type, depending on the specific genetic background or presence/absence of specific coexisting biochemical defects [33].
Future research will have to address these mechanisms in more detail and is required to determine whether the growth-suppressing effect of LRP1B extends from the inhibition of tumor cell growth to a possible role of LRP1B in other cells, as e.g. the formation of atherosclerotic lesions.
MATERIALS AND METHODS
Cloning of full-length murine Lrp1b
Total RNA was prepared from mouse brain (C57black6/J) using RNAStat (Amsbio) and 1μg transcribed into cDNA using Oligo dT12-18 (Invitrogen) and SuperScript® II Reverse Transcriptase (Clontech) as suggested by the manufacturer. Preparative PCR was performed with Takara-brand LA PCR Kit, Version 2.1 (Clontech) according to the manufacturer’s instructions.
Restriction enzymes were purchased either from NEB, Fermentas or Roche and used as recommended by the manufacturer (with 5-fold amount of enzyme for cleaving nonlinear DNA).
Plasmids were purchased from Invitrogen (pcDNA3.1TM(-)/myc-His A and pCR2.1 Topo, the latter being part of the TA Cloning Kit). DNA concentrations were determined photometrically. For agarose gel electrophoresis SeaKem® LE Agarose (Lonza) was used at appropriate concentrations containing GelRedTM Nucleic Acid Gel Stain (Biotium) for analytic gels, and SeaKem® GTG Agarose (Lonza) for preparative gels, respectively.
TaKaRa DNA Ligation Kit Long (Clontech) and DNA ligation kit version 2 (Takara) were used for ligation reactions as suggested including appropriate negative and positive controls. TaKaRa DNA Ligation Kit Long was used for ligations involving fragments longer then 10kB (Ligation for 15h at 16°C). Subcloning of PCR fragments into the pCR2.1 Topo TA vector was performed as suggested by the manufacturer.
For transformation reactions XL10-Gold Ultracompetent Cells purchased from Agilent technologies (for longer fragments) or Library efficient DH5a competent cells (Gibco) were used. Selection was done on LB agar plates containing 100mg/l Ampicillin (Sigma). Clones were picked and grown in LB medium containing 100mg/l ampicillin overnight. Plasmid DNA was prepared using QIAprep Spin Miniprep Kit (Qiagen).
Large scale plasmid preparation was done using Qiagen Plasmid Maxi Kit (Qiagen). Digestion was performed as suggested by the manufacturer. For preparative digestions, an aliquot of the reaction was removed after 3 hours and subjected to gel electrophoresis for verification of complete digestion.
Sequences of all used primers are given in Supplementary Table S1.
Cell lines and cell culture
Human embryonal kidney (HEK) 293 cells and Calu-1 NSCLC cells were purchased from the American Type Culture Collection (ATCC) and cultured in Dulbecco’s modified Eagle medium DMEM (Lonza) supplemented with 10 % fetal calf serum (FCS) (Sigma Aldrich), 4mM glutamine (PAA Laboratories) and 1% penicillin/streptomycin (PAA Laboratories). NSCLC cell lines A427, A549 and HCC-44 were purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ) and cultured in RPMI 1640 (Lonza) supplemented with 10 % fetal calf serum (FCS), 4mM glutamine and 1% penicillin/streptomycin.
Transfection of HEK 293 cells with the full-length mLrp1b expression vector
HEK 293 cells were grown to 80-90% confluency. After changing the medium (DMEM) to antibiotic free medium, FuGENE 6 transfection reagent (Promega) was used according to the manufacturer’s instructions. Briefly, for a 25 cm2 flask with 2 ml medium, 100μl serum free medium plus 6μl Fugene and 2μg of linearized plasmid were used for the transfection mix. After 6 hours the transfected cells were selected and maintained in the presence of G418 (Gibco) at 0.4 mg/ml.
Overexpression and knockdown of LRP1B in non-small lung cancer cells
A427, A549 amd HCC-44 cells were stably transfected with the generated full-length Lrp1b vector, a plasmid coding for a human LRP1B region IV minireceptor [19] or an empty vector as control, respectively, using FuGENE 6 as described above.
To knock down endogenous LRP1B expression, Calu-1 cells were transfected with 50 nM SMARTpool: ON-TARGETplus LRP1B siRNA (Dharmacon) or scrambled (SCR) control siRNA (ON-TARGETplus non-targeting pool; Dharmacon) using DharmaFect 1 transfection reagent (Dharmacon) according to manufacturer’s instructions.
RNA isolation, reverse transcription and quantitative real-time PCR
Total RNA was extracted from whole cells using RNeasy Mini Kit (Qiagen) according to manufacturer’s instructions and reverse-transcribed with M-MLV reverse transcriptase (Promega). Quantitative PCR (qPCR) was performed using the SsoFast™ EvaGreen® Supermix (BioRad) and the CFX96 PCR System (BioRad) with CFX96 Manager software on a Windows 7 platform according to manufacturer’s instructions. The specificity of PCR products was confirmed by melting curve analysis. cDNA concentrations were normalized to the internal standard ribosomal protein, large, P0 (RPLP0), a moderate copy number housekeeping gene not regulated under the experimental conditions used. Fold change in gene expression was determined using the mathematical model ratio 2-ΔΔCT [34]. Primers were designed using Primer3Plus [35] to allow specific quantification and differentiation of human LRP1B (endogenous receptor, LRP1B minireceptor) and mouse Lrp1b (full-length recombinant Lrp1b receptor) sequences. Sequences of all used primers are given in Supplementary Table S1.
Western blot analysis
Total cell extracts were prepared as described previously [36] and were separated with 4-15% reducing SDS gels followed by immunoblotting with an affinity-purified, polyclonal anti-LRP1B antibody raised against the 13 C-terminal amino acids (identical in human LRP1B and mouse Lrp1b) as previously described [19]. Crude membrane preparations from mouse brain were prepared as described [19] and used as positive control.
Proliferation assay
Relative quantification of DNA synthesis was determined by bromodeoxyuridine (BrdU) incorporation as described previously [37]. Briefly, stably transfected NSCLC cells or Calu-1 cells 72 h post-transfection with siRNA were incubated at a density of 5000/well in 96 well plates in culture medium containing 1% FCS. Proliferation rate after 72 h was analyzed by the Cell Proliferation ELISA BrdU Kit (Roche Applied Science) according to manufacturer’s instructions.
Identification of intracellular signaling pathways
A427 cells stably transfected with plasmids containing either murine full-length Lrp1b or empty vector (EV) control were serum-starved overnight and subsequently stimulated with 10% fetal calf serum. Total cell extracts from these cells were analyzed using the following antibodies: Phospho-Akt (Cell Signaling Technology #9271); Phospho-p44/42 MAPK (Erk1/2; Cell Signaling Technology #9101); p21CIP1 (BD Pharmingen #556430); Phospho-Src (Tyr416; Cell Signaling Technology #2101); Phospho-Src (Tyr527; Cell Signaling Technology #2105); Phospho-Stat3 (Ser727; Cell Signaling Technology #9134); Phospho-Stat3 (Tyr705; Cell Signaling Technology Antibody #9131); GAPDH (Abcam #ab37187).
Statistics
Numeric data are presented as mean ± SEM from at least three independent experiments. Statistical differences between treatments were calculated using Student’s paired t-test.
ACKNOWLEDGMENTS
We thank Dr. K. Salzmann and U. Stanzl for valuable help with some experiments.
CONFLICTS OF INTEREST
The authors report no conflicts of interest.
GRANT SUPPORT
Supported by grant P 20825-B05 of the Austrian Science Fund (FWF) to P.M.
REFERENCES
1. Liu CX, Musco S, Lisitsina NM, Yaklichkin SY, Lisitsyn NA. Genomic organization of a new candidate tumor suppressor gene, LRP1B. Genomics. 2000; 69: 271–4.
2. Langbein S, Szakacs O, Wilhelm M, Sukosd F, Weber S, Jauch A, Lopez Beltran A, Alken P, Kälble T, Kovacs G. Alteration of the LRP1B gene region is associated with high grade of urothelial cancer. Lab Invest. 2002; 82: 639–43.
3. Pineau P, Marchio A, Nagamori S, Seki S, Tiollais P, Dejean A. Homozygous deletion scanning in hepatobiliary tumor cell lines reveals alternative pathways for liver carcinogenesis. Hepatology. 2003; 37: 852–61.
4. Sonoda I, Imoto I, Inoue J, Shibata T, Shimada Y, Chin K, Imamura M, Amagasa T, Gray JW, Hirohashi S, Inazawa J. Frequent silencing of low density lipoprotein receptor-related protein 1B (LRP1B) expression by genetic and epigenetic mechanisms in esophageal squamous cell carcinoma. Cancer Res. 2004; 64: 3741–7.
5. Hirai Y, Utsugi K, Takeshima N, Kawamata Y, Furuta R, Kitagawa T, Kawaguchi T, Hasumi K, Noda T, Noda ST. Putative gene loci associated with carcinogenesis and metastasis of endocervical adenocarcinomas of uterus determined by conventional and array-based CGH. Am J Obstet Gynecol. 2004; 191: 1173–82.
6. Roversi G, Pfundt R, Moroni RF, Magnani I, van Reijmersdal S, Pollo B, Straatman H, Larizza L, Schoenmakers, E F P M. Identification of novel genomic markers related to progression to glioblastoma through genomic profiling of 25 primary glioma cell lines. Oncogene. 2006; 25: 1571–83.
7. Nakagawa T, Pimkhaokham A, Suzuki E, Omura K, Inazawa J, Imoto I. Genetic or epigenetic silencing of low density lipoprotein receptor-related protein 1B expression in oral squamous cell carcinoma. Cancer Sci. 2006; 97: 1070–4.
8. Rahmatpanah FB, Carstens S, Guo J, Sjahputera O, Taylor KH, Duff D, Shi H, Davis JW, Hooshmand SI, Chitma-Matsiga R, Caldwell CW. Differential DNA methylation patterns of small B-cell lymphoma subclasses with different clinical behavior. Leukemia. 2006; 20: 1855–62.
9. Taylor KH, Pena-Hernandez KE, Davis JW, Arthur GL, Duff DJ, Shi H, Rahmatpanah FB, Sjahputera O, Caldwell CW. Large-scale CpG methylation analysis identifies novel candidate genes and reveals methylation hotspots in acute lymphoblastic leukemia. Cancer Res. 2007; 67: 2617–25.
10. Lu Y, Wu C, Li H, Liu H, Lu C, Leu Y, Wang C, Chen L, Lin K, Chang Y. Aberrant methylation impairs low density lipoprotein receptor-related protein 1B tumor suppressor function in gastric cancer. Genes Chromosomes Cancer. 2010; 49: 412–24.
11. Prazeres H, Torres J, Rodrigues F, Pinto M, Pastoriza MC, Gomes D, Cameselle-Teijeiro J, Vidal A, Martins TC, Sobrinho-Simões M, Soares P. Chromosomal, epigenetic and microRNA-mediated inactivation of LRP1B, a modulator of the extracellular environment of thyroid cancer cells. Oncogene. 2011; 30: 1302–17.
12. Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, Harshman K, Guipponi M, Bukach O, Zoete V, Michielin O, Muehlethaler K, Speiser D, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet. 2012; 44: 133–9.
13. Cowin PA, George J, Fereday S, Loehrer E, van Loo P, Cullinane C, Etemadmoghadam D, Ftouni S, Galletta L, Anglesio MS, Hendley J, Bowes L, Sheppard KE, et al. LRP1B deletion in high-grade serous ovarian cancers is associated with acquired chemotherapy resistance to liposomal doxorubicin. Cancer Res. 2012; 72: 4060–73.
14. Ni S, Hu J, Duan Y, Shi S, Li R, Wu H, Qu Y, Li Y. Down expression of LRP1B promotes cell migration via RhoA/Cdc42 pathway and actin cytoskeleton remodeling in renal cell cancer. Cancer Sci. 2013; 104: 817–25.
15. Ross JS, Wang K, Rand JV, Gay L, Presta MJ, Sheehan CE, Ali SM, Elvin JA, Labrecque E, Hiemstra C, Buell J, Otto GA, Yelensky R, et al. Next-generation sequencing of adrenocortical carcinoma reveals new routes to targeted therapies. J Clin Pathol. 2014; 67: 968–73.
16. Ding D, Lou X, Hua D, Yu W, Li L, Wang J, Gao F, Zhao N, Ren G, Li L, Lin B. Recurrent targeted genes of hepatitis B virus in the liver cancer genomes identified by a next-generation sequencing-based approach. PLoS Genet. 2012; 8: e1003065.
17. Hu Z, Zhu D, Wang W, Li W, Jia W, Zeng X, Ding W, Yu L, Wang X, Wang L, Shen H, Zhang C, Liu H, et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat Genet. 2015; 47: 158–63.
18. Tanaga K, Bujo H, Zhu Y, Kanaki T, Hirayama S, Takahashi K, Inoue M, Mikami K, Schneider WJ, Saito Y. LRP1B attenuates the migration of smooth muscle cells by reducing membrane localization of urokinase and PDGF receptors. Arterioscler Thromb Vasc Biol. 2004; 24: 1422–8.
19. Marschang P, Brich J, Weeber EJ, Sweatt JD, Shelton JM, Richardson JA, Hammer RE, Herz J. Normal development and fertility of knockout mice lacking the tumor suppressor gene LRP1b suggest functional compensation by LRP1. Mol Cell Biol. 2004; 24: 3782–93.
20. Dietrich MF, van der Weyden, Louise, Prosser HM, Bradley A, Herz J, Adams DJ. Ectodomains of the LDL receptor-related proteins LRP1b and LRP4 have anchorage independent functions in vivo. PLoS ONE. 2010; 5: e9960.
21. Liu CX, Li Y, Obermoeller-McCormick LM, Schwartz AL, Bu G. The putative tumor suppressor LRP1B, a novel member of the low density lipoprotein (LDL) receptor family, exhibits both overlapping and distinct properties with the LDL receptor-related protein. J Biol Chem. 2001; 276: 28889–96.
22. Haas J, Beer AG, Widschwendter P, Oberdanner J, Salzmann K, Sarg B, Lindner H, Herz J, Patsch JR, Marschang P. LRP1b shows restricted expression in human tissues and binds to several extracellular ligands, including fibrinogen and apoE-carrying lipoproteins. Atherosclerosis. 2011; 216: 342–7.
23. Herz J, Hamann U, Rogne S, Myklebost O, Gausepohl H, Stanley KK. Surface location and high affinity for calcium of a 500-kd liver membrane protein closely related to the LDL-receptor suggest a physiological role as lipoprotein receptor. EMBO J. 1988; 7: 4119–27.
24. Li Y, Knisely JM, Lu W, McCormick LM, Wang J, Henkin J, Schwartz AL, Bu G. Low density lipoprotein (LDL) receptor-related protein 1B impairs urokinase receptor regeneration on the cell surface and inhibits cell migration. J Biol Chem. 2002; 277: 42366–71.
25. Herz J, Marschang P. Coaxing the LDL receptor family into the fold. Cell. 2003; 112: 289–92.
26. Knisely JM, Li Y, Griffith JM, Geuze HJ, Schwartz AL, Bu G. Slow endocytosis of the LDL receptor-related protein 1B: implications for a novel cytoplasmic tail conformation. Exp Cell Res. 2007; 313: 3298–307.
27. Willnow TE, Orth K, Herz J. Molecular dissection of ligand binding sites on the low density lipoprotein receptor-related protein. J Biol Chem. 1994; 269: 15827–32.
28. Kreiling JL, Byrd JC, MacDonald RG. Domain interactions of the mannose 6-phosphate/insulin-like growth factor II receptor. J Biol Chem. 2005; 280: 21067–77.
29. Liu C, Ranganathan S, Robinson S, Strickland DK. gamma-Secretase-mediated release of the low density lipoprotein receptor-related protein 1B intracellular domain suppresses anchorage-independent growth of neuroglioma cells. J Biol Chem. 2007; 282: 7504–11.
30. Singhal S, Vachani A, Antin-Ozerkis D, Kaiser LR, Albelda SM. Prognostic implications of cell cycle, apoptosis, and angiogenesis biomarkers in non-small cell lung cancer: a review. Clin Cancer Res. 2005; 11:3974-86.
31. Dehm SM, Bonham K. SRC gene expression in human cancer: the role of transcriptional activation. Biochem Cell Biol. 2004; 82: 263–74.
32. Lillis AP, Van Duyn, Lauren B, Murphy-Ullrich JE, Strickland DK. LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol Rev. 2008; 88: 887–918.
33. Zhang H, Lai R. STAT3 in Cancer-Friend or Foe? Cancers (Basel). 2014; 6: 1408–40.
34. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25: 402–8.
35. Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3--new capabilities and interfaces. Nucleic Acids Res. 2012; 40: e115.
36. Zenzmaier C, Untergasser G, Hermann M, Dirnhofer S, Sampson N, Berger P. Dysregulation of Dkk-3 expression in benign and malignant prostatic tissue. Prostate. 2008; 68: 540–7.
37. Zenzmaier C, Sampson N, Pernkopf D, Plas E, Untergasser G, Berger P. Attenuated proliferation and trans-differentiation of prostatic stromal cells indicate suitability of phosphodiesterase type 5 inhibitors for prevention and treatment of benign prostatic hyperplasia. Endocrinology. 2010; 151: 3975–84.