
INTRODUCTION
Chronic inflammation plays an important role in cancer development [1]. In particular, colorectal carcinoma (CRC) is strongly associated with an inflammatory microenvironment and used as a model of the link between chronic inflammation and cancer. Many studies have shown that inflammatory responses promote intestinal carcinogenesis by altering the intestinal microenvironment [2]. Indeed, experimental colitis enhances intestinal carcinogenesis in ApcMin/+ mice through an inducible nitric oxide synthase (iNOS)-dependent mechanism [3]. As Apc gene mutations are responsible for the familial adenomatous polyposis, and more than 80% of CRC harbor mutations in APC, the ApcMin/+ mice model is considered a relevant intestinal carcinogenesis model [4–5].
Nuclear factor (NF)-κB is a family of transcription factors that regulate the expression of genes involved in cell growth and survival, stress response, and inflammation [6]. For example, urokinase-type plasminogen activator (uPA); vascular endothelial growth factor; and some of the pro-inflammatory factors, such as interleukin-1 (IL-1), tumor necrosis factor (TNF)-α, and iNOS, are regulated by NF-κB [7]. In mammals, NF-κB is composed of RelA (p65), RelB, c-Rel, NF-κB1 (p105/P50), and NF-κB2 (p100/P52). These proteins form homo- or heterodimers. Among them, RelA is the most important subunit of the NF-κB family. Recently, it has become clear that NF-κB signaling plays important roles in the development and progression of cancer [8–10]. NF-κB controls the ability of both preneoplastic and malignant cells to resist apoptosis. It may also regulate tumor angiogenesis and invasiveness. Regarding the effect of NF-κB signaling in colon cancer, significant evidence suggests that NF-κB signaling plays a role in colorectal cancer promotion [2, 11]. Moreover, the activation of NF-κB is reported in over 50% of CRCs [12], suggesting that NF-κB signaling may be a promising therapeutic target in CRCs. Dehydroxymethylepoxyquinomicin (DHMEQ) is a synthetic small molecule that inhibits nuclear translocation of RelA. Previous studies showed that DHMEQ inhibits various types of inflammation and cancer cell growth [13–26].
HAI-1 is a membrane-associated Kunitz-type serine protease inhibitor that is expressed on most epithelial cells and placental cytotrophoblasts [27]. We have previously reported that intestinal barrier function was decreased in intestine-specific HAI-1-deficient mice, and these mice showed increased susceptibility of DSS-induced colitis [28]. Moreover, the loss of HAI-1 accelerated tumor formation both in an inflammation-associated mice colon carcinogenesis model and in a mutant Apc model [29]. However, the molecular mechanism of increased tumor formation in HAI-1-deficient mice is still unclear. Because it was reported that mucosal damage plays a role in the pathogenesis of colitis and CRC [12], we speculated that in the HAI-1-deficient intestine, the inflammatory response was induced and NF-κB signaling was activated, which involved HAI-1 loss-induced acceleration of tumor formation.
In this study, we analyzed the effect of DHMEQ administration on intestinal tumor formation to clarify whether the acceleration of intestinal carcinogenesis observed in HAI-1-deficient mice is associated with the activation of NF-κB signaling or not. We found that NF-κB was activated in HAI-1-deficient ApcMin/+ mice tumors. Moreover, inhibition of NF-κB by DHMEQ treatment significantly reduced tumor formation in HAI-1-deficient ApcMin/+ mice. These results suggest that HAI-1 has a suppressive role in intestinal carcinogenesis by regulating of NF-κB signaling.
RESULTS
NF-κB signaling was activated in HAI-1-deleted intestine
We have previously reported that the loss of HAI-1 in intestinal epithelial cells altered intestinal barrier function and increased susceptibility to experimental colitis [28]. Therefore, we hypothesized that NF-κB signaling is activated in the HAI-1-deficient intestine. To confirm this hypothesis, we evaluated the nuclear translocation of RelA/p65, the subunit of NF-κB, using Spint1LoxP/LoxP/Villin-Cre+/0/ ApcMin/+ mice (hereafter HAI-1-deficient ApcMin/+ mice) at 15 weeks of age, the same mice as in our previous study [29]. The defect of HAI-1 protein was confirmed by immunohistochemistry (Supplementary Figure S1). In normal mucosa of the small intestine, an increased number of nuclear RelA/p65-positive cells were observed in both epithelial and stromal components of HAI-1-deficient ApcMin/+ mucosa compared to control ApcMin/+ mucosa (Figure 1A). In tumor tissues, the nuclear translocation of RelA/p65 was also increased in both tumor cells and stromal cells of the HAI-1-deficient intestine (Figure 1B). We also examined the expression of NF-κB target genes, such as TNF-α, IL-6, and uPA. In tumor tissue, the expression of TNF-α and IL-6 was increased both in control mice and HAI-1-deficient mice. However, in the non-tumor mucosa, the expression levels of these genes were significantly increased in HAI-1-deficient mice compared with control mice (Figure 1C, 1D). The uPA expression was upregulated both in non-tumor mucosa and tumor tissue in HAI-1 deficient mice (Figure 1C, 1D).
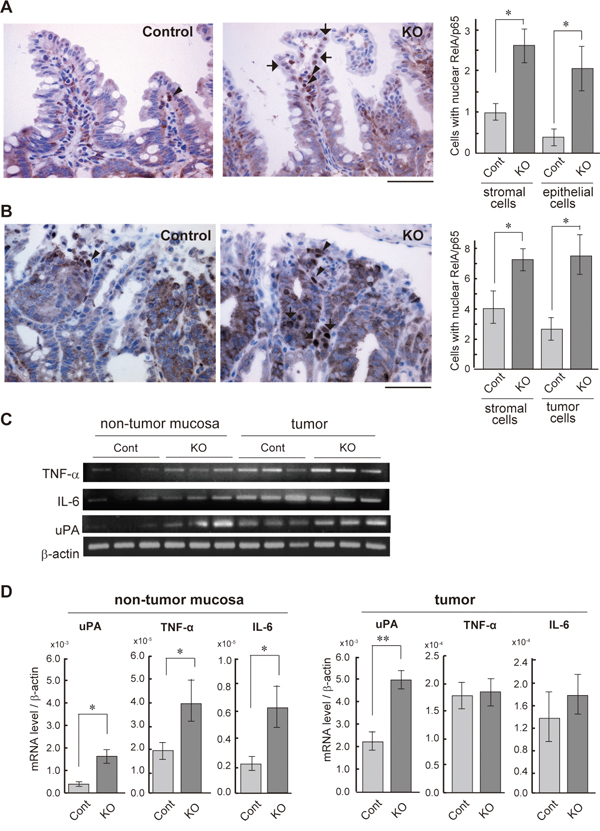
Figure 1: NF-κB activation status of the HAI-1-deficient intestine. (A) Immunohistochemical analyses of RelA/p65 in the non-tumor mucosa of small intestine from control (Cont; n = 4) and HAI-1-deficient (KO; n = 4) ApcMin/+ mice at 15 weeks of age. Arrows and arrowheads indicate the nuclear RelA/p65-positive epithelial cells and stromal cells, respectively. Positive cells/200× field were counted and plotted in the right panel (mean ± SEM). *, p < 0.05. Bar, 50 μm. (B) Nuclear immunoreactivity of RelA/p65 in intestinal tumor tissue. Arrows and arrowheads indicate the nuclear RelA/p65-positive tumor cells and stromal cells, respectively. Right panel indicates mean number ± SEM of positive cells/200× field. *, p < 0.05. Bar, 50 μm. (C) RT-PCR of NF-κB target genes in non-tumor mucosa and tumor tissues of control and KO mice. (D) Quantitative RT-PCR for mRNAs of NF-κB target genes. *, p < 0.05, **, p < 0.005.
NF-κB inhibitor alleviated the HAI-1 loss-induced enhanced formation of intestinal tumors
We then investigated the effect of DHMEQ on tumor formation in HAI-1-deficient ApcMin/+ mice. The mice were treated with an intraperitoneal administration of saline with or without DHMEQ (three times per week for 4 weeks) and sacrificed 12 weeks after birth. Ten weeks after birth, the body weight gain of HAI-1-deficient ApcMin/+ mice was lower than control mice. However, the intraperitoneal administration of DHMEQ alleviated body weight loss in HAI-1-deficient ApcMin/+ mice (Figure 2A). It is well known that ApcMin/+ mice exhibit anemia accompanied by increased tumor formation [30] and our previous studies revealed that HAI-1-deficient ApcMin/+ mice developed severe anemia compared with control mice [29]. In the present study, blood hemoglobin concentrations were also improved after DHMEQ treatment in HAI-1-deficient ApcMin/+ mice (Figure 2B). At 12 weeks of age, the number of intestinal tumors was significantly increased in HAI-1-deficient ApcMin/+ mice (116 ± 8.0, n = 5) compared with control mice (46.7 ± 8.0, n = 6) (Figure 2C). Interestingly, DHMEQ treatment significantly reduced the number of intestinal tumors (63.5 ± 3.0, n = 6) in HAI-1-deficient ApcMin/+ mice to a level comparable with that in control ApcMin/+ mice (Figure 2C). On the other hand, the administration of DHMEQ had no effect on body weight gain, hemoglobin concentrations, and tumor formation in control mice (Figure 2A-2C). DHMEQ treatment tended to decrease the sizes of tumors both in HAI-1-deficient ApcMin/+ mice and control mice. However, the differences were not significant (Figure 2C). Tumor histology was similar between the two groups with or without DHMEQ treatment (Figure 2D). We also examined the effect of DHMEQ on nuclear translocation of RelA/p65 in the tumor tissues of HAI-1-deficient ApcMin/+ mice. As expected, administration of DHMEQ significantly suppressed the nuclear translocation of RelA/p65 both in tumor and stromal cells of HAI-1-deficient ApcMin/+ mice (Figure 3A). However, DHMEQ did not affect Wnt signaling, as judged by immunohistochemical analysis of β-catenin (Figure 3B). While nuclear β-catenin-positive tumor cells were modestly increased in the intestinal polyps from HAI-1-deficient ApcMin/+ mice compared to those from control ApcMin/+ mice, the nuclear translocation of β-catenin was not alleviated by DHMEQ treatment in both tumors. In addition, the expression level of APC was not altered by the HAI-1 deficiency (Supplementary Figure S2).
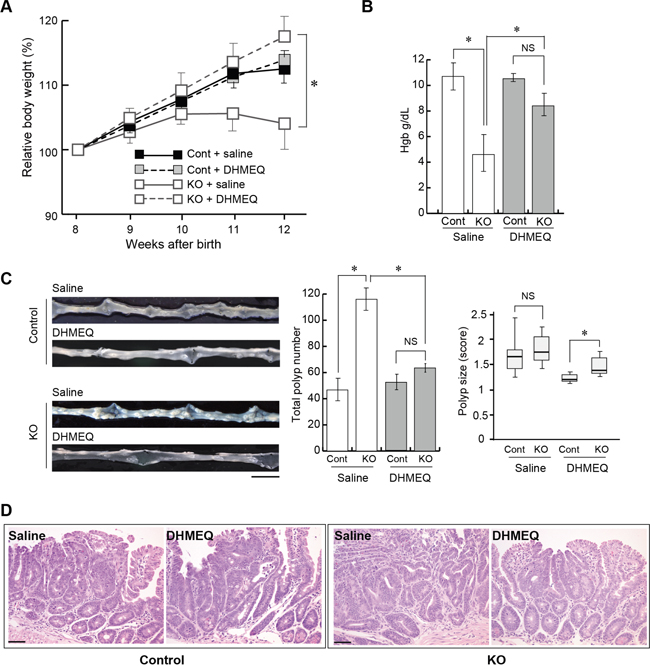
Figure 2: Effect of DHMEQ on intestinal tumorigenesis in HAI-1-deficient ApcMin/+ mice. (A) Relative body weight (mean ± SEM) of Cont (saline; n = 6), Cont (DHMEQ; n = 5), KO (saline; n = 5), and KO (DHMEQ; n = 6) groups. Mean body weight at the start point (8 weeks of age) of each group was 21.47 (Cont + saline), 21.42 (Cont + DHMEQ), 21.18 (KO + saline) or 20.33 (KO + DHMEQ), without statistically significant differences between groups. *, p < 0.05. (B) Mean hemoglobin concentration (mean ± SEM) of the mice. NS, not significant; *, p < 0.05. (C) Representative macroscopic appearance of the small intestine from saline- or DHMEQ-treated control and KO mice (left panel), the number of intestinal polyps (mean ± SEM) of the mice (middle panel), and polyp size scores of the mice (right panel). The box shows the interquartile range, and the whiskers indicate the sample maximum and minimum. The median is indicated by a bold line. *, p < 0.05. Bar, 1 cm. (D) Histology of tumors (H&E stain). Bar, 50 μm.
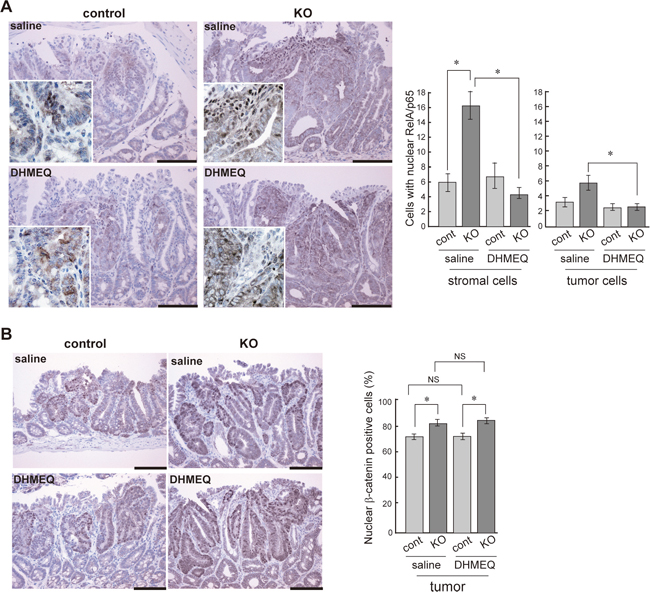
Figure 3: Effect of DHMEQ on nuclear translocation of RelA/p65 and β-catenin in HAI-1-deficient ApcMin/+ mice tumor. Immunostaining of RelA/p65 (A), and β-catenin (B) of control and KO tumor before and after DHMEQ treatment. The enlarged image is also shown as an inset. Bar, 100 μm. *, p < 0.01. NS, not significant.
DISCUSSION
In this study, we showed that loss of HAI-1 in the intestine leads to activation of NF-κB signaling. Treatment with DHMEQ, a synthesized small molecule inhibitor of NF-κB, alleviated the HAI-1 loss-induced acceleration of tumor formation in Apc mutant mice. Consequently, DHMEQ improved health parameters, such as body weight and hemoglobin concentration, in HAI-1-deficient ApcMin/+ mice. This study also revealed that DHMEQ has no effect on tumor incidence in control mice. Because it has been reported that activation of NF-κB signaling in intestinal epithelial cells and lamina propria macrophages play a major role in the development of colitis-associated cancer (CAC) in humans [11], this study indicates that DHMEQ or its derivatives may have an implication in CRC prevention and therapy.
Although the precise mechanisms underlying the HAI-1 loss-induced activation of NF-κB signaling remain unclear, some likely mechanisms are suggested based on the proposed function of proteases regulated by HAI-1. One is HGF-mediated activation of NF-κB signaling. HAI-1 is a potent inhibitor of pro-HGF-activating proteases, such as HGF activator, matriptase, hepsin, TMPRSS13, and human airway trypsin-like protease (HAT) [31], and our previous study demonstrated that enhanced HGF activation is observed in tumor tissue and non-tumor mucosa of HAI-1-deficient ApcMin/+ mice [29]. HGF is known to have important roles in cancer progression through its specific receptor, MET [31]. Furthermore, it has been reported that HGF/MET signaling activates a variety of signaling pathways, such as PI3K/AKT, ERK, NF-κB, and STAT3 [32, 33]. Another candidate for activating NF-κB signaling is uPA. uPA and its receptor uPAR are known to have a tumor-promoting role and are involved in cell processes such as migration, proliferation, and angiogenesis [34]. Previous studies revealed that uPA increases nuclear NF-κB p65 levels in meningioma cells and neutrophils [33, 34]. uPA is produced by cancer cells and stromal cells as a proenzyme uPA (pro-uPA) and is activated in the tumor microenvironment [36]. Similar to pro-HGF, pro-uPA is a substrate for HAI-1-targeted serine proteases, such as matriptase and hepsin [37]. Moreover, expression of uPA was significantly increased both in non-tumor mucosa and tumor tissue in HAI-1-deficient ApcMin/+ mice (Figure 1C, 1D). Therefore, uPA may have a role in the activation of NF-κB signaling in HAI-1-deficient ApcMin/+ mice. Protease-activated receptor 2 (PAR-2) signaling may also be involved in NF-κB activation. PAR-2 is expressed by intestinal epithelial cells, and it is well known that trypsin-like serine proteases regulated by HAI-1, such as matriptase and prostasin, and kallikrein-5 (KLK5) activate PAR-2 [38]. PAR-2 activation induces various intracellular signalings, such as p38 mitogen-activated protein kinase [39], Rho [40], and NF-κB signaling [41–43]. A recent study showed that PAR-2 activation by matriptase induces NF-κB activity that leads to the release of pro-tumorigenic inflammatory cytokines in a chemically-induced skin carcinogenesis model [43]. Another evidence is that KLK5-mediated activation of PAR-2 in oral squamous cell carcinoma leads to the activation of NF-κB signaling, which increases expression of inflammatory cytokines and suppresses anti-inflammatory tumor suppressor microRNAs [42]. These studies suggested that in HAI-1-deficient ApcMin/+ mice intestine, PAR-2 was activated by HAI-1 targeted serine proteases, which may cause the activation of NF-κB signaling. Indeed, the activity of trypsin-like serine protease was significantly increased in HAI-1-deficient ApcMin/+ mice mucosa (Supplementary Figure S2). While matriptase, a major target of HAI-1 with an efficient PAR-2 activating activity, was expressed in the intestinal mucosa, its mRNA level was not altered by the defect of HAI-1 (Supplementary Figure S2). At present, it remains uncertain whether the increased trypsin-like protease activity in HAI-1-deficient mucosa is caused by dysregulated matriptase or other serine proteases. Taken together, excess activation of HGF, uPA, and/or PAR-2 due to the absence of HAI-1 may induce the activation of NF-κB signaling and produce pro-tumorigenic inflammatory cytokines that may be responsible for increased intestinal tumorigenesis. Further study will be required to clarify the detailed mechanisms underlying the increased intestinal tumorigenesis in HAI-1-deficient ApcMin/+ mice.
In summary, we demonstrated that treatment with DHMEQ, a small molecule inhibitor of NF-κB, reduced tumor formation and improved the health condition without adverse side effects in HAI-1-deficient ApcMin/+ mice. This suggests that DHMEQ serves as a promising agent for the treatment of NF-κB-activated colon cancer.
MATERIALS AND METHODS
Mice
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Miyazaki. HAI-1(Spint1)-deficient ApcMin/+ mice were established by crossing Spint1LoxP/LoxP mice with mice harboring the Cre recombinase under the control of villin promoter and ApcMin/+ mice (Spint1LoxP/LoxP/Villin-Cre+/0/ ApcMin/+) [29]. Spint1LoxP/LoxPApcMin/+ mice were used as a control.
Murine intestinal tumorigenesis models
ApcMin/+ mice were used for analyzing the effect of DHMEQ, a small molecule inhibitor of NF-κB. Eight-week-old male HAI-1/Spint1-deficient and control ApcMin/+ mice were injected intraperitoneally with 10 mg/kg DHMEQ three times per week and were sacrificed 12 weeks after birth and autopsied to evaluate the number and sizes of tumors formed. Scoring of tumor size was calculated as previously described [29]. For histological analysis, intestinal tissues were fixed in 4% formaldehyde in phosphate-buffered saline (PBS) and embedded in paraffin. Four-micrometer-thick sections were stained with hematoxylin and eosin (HE).
RT-PCR
Total RNA was prepared with TRIzol™ (Life Technologies Japan, Tokyo, Japan), followed by DNase I (Takara Bio, Shiga, Japan) treatment. For RT-PCR, 3 μg of total RNA was reverse-transcribed with a mixture of Oligo (dT)12-18 (Life Technologies Japan) and random primers (6mers) (Takara Bio) using 200 units of ReverTra Ace™ (TOYOBO, Osaka, Japan), and 1/30th of the resulting cDNA was processed for each PCR reaction with 0.2 μM of both forward and reverse primers and AmpliTaq Gold™ PCR Master Mix (Life Technologies Japan). The thermal cycle profile was 15 sec at 94°C, 15 sec at 60°C, and 1 min at 70°C. The primer sequences for β-actin, TNFα, IL-6, uPA, matriptase and APC are described in Table 1. For qRT-PCR, PCR was performed in a Thermal Cycler Dice Real Time System II (Takara Bio) using the SYBR Premix Ex Taq II (Takara Bio). For internal control, β-actin mRNA was also measured.
Table 1: Primer for RT-PCR
Gene |
Forward |
Reverse |
size |
|---|---|---|---|
β-actin |
5’-TGACAGGATGCAGAAGGAGA |
5’-GCTGGAAGGTGGACAGTGAG |
131 |
TNF-α |
5’-TCGAGTGACAAGCCTGTAGC |
5’-GGAGGTTGACTTTCTCCTGG |
255 |
IL-6 |
5’-AGCCAGAGTCCTTCAGAGAG |
5’-ACTCCTTCTGTGACTCCAGC |
136 |
uPA |
5’-GAAGTTTGAGGTGGAGCAGC |
5’-CCCGTGCTGGTACGTATCTT |
101 |
Matriptase |
5’-CACGAATGATGTGTGTGGGTTTC |
5’-CCTGGAACATTCGCCCATCT |
105 |
APC |
5’-TGTCTGCACACTGCACTGAG |
5’-CTTGCTGAGAGATTCCAC |
286 |
Immunohistochemical analyses
Intestinal tissues of HAI-1-deficient ApcMin/+ mice and control mice were fixed in 4% paraformaldehyde in PBS overnight and then dehydrated and embedded in paraffin. For immunohistochemistry, tissue sections were processed for antigen retrieval by microwaving for 10 min at 96°C in 10 mM citrate buffer (pH 6.0), followed by treatment with 3% H2O2 in PBS for 10 min. After blocking in 5% normal goat serum (DAKO, Glostrup, Denmark) in PBS, the sections were incubated with anti-NF-κB p65 rabbit monoclonal antibody (Cell Signaling Technology, Tokyo, Japan) or anti-β-catenin rabbit polyclonal antibody (Sigma-Aldrich, St. Louis, MO) or anti-HAI–1 goat polyclonal antibody (R&D Systems, Minneapolis, MN) for 16 h at 4°C and then incubated with Envision™ labeled polymer reagents (DAKO) for 30 min at room temperature. The reactions were revealed by nickel, cobalt-3, 3′-diaminobenzidine (Pierce, Rockford, IL) and counterstained with Mayer’s hematoxylin. To quantify the nuclear translocation of NF-κB or β-catenin, ten randomly selected areas were photographed at 200× magnification in both normal (non-tumor) intestinal mucosal tissue and tumor tissue from each mouse. Then, two independent investigators counted the nuclear RelA or β-catenin-positive cells and the mean number per field were calculated.
Statistical analysis
Statistical analysis was performed using SPSS15.0 software (SPSS Japan Inc., Tokyo, Japan). Comparison between two unpaired groups was made with the Mann-Whitney U test. Significance was set at p < 0.05.
ACKNOWLEDGMENTS
We thank Ms. Yumiko Nomura and Yukari Torisu for their excellent technical assistance. This work was supported by Grant-in-Aid for Scientific Research no.15K08311 (M. Kawaguchi) and no.24390099 (H. Kataoka) from the Ministry of Education, Science, Sports and Culture, Japan.
CONFLICTS OF INTEREST
The authors declare that there is no conflicts of interest.
REFERENCES
1. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010; 140:883-899.
2. Carothers AM, Davids JS, Damas BC, Bertagnolli MM. Persistent Cyclooxygenase-2 Inhibition Downregulates NF- B, Resulting in Chronic Intestinal Inflammation in the Min/+ Mouse Model of Colon Tumorigenesis. Cancer Res. 2010; 70:4433-4442.
3. Kohno H, Takahashi M, Yasui Y, Suzuki R, Miyamoto S, Kamanaka Y, Naka M, Maruyama T, Wakabayashi K, Tanaka T. A specific inducible nitric oxide synthase inhibitor, ONO-1714 attenuates inflammation-related large bowel carcinogenesis in male Apc(Min/+) mice. Int J Cancer. 2007; 121:506-513.
4. Wang, L.; Zhang, Q. Application of the Apc(Min/+) mouse model for studying inflammation-associated intestinal tumor. Biomed. Pharmacother. 2015; 71, 216-221.
5. Altamemi, I.; Murphy, E. A.; Catroppo, J. F.; Zumbrun, E. E.; Zhang, J.; McClellan, J. L.; Singh, U. P.; Nagarkatti, P. S.; Nagarkatti, M. Role of microRNAs in resveratrol-mediated mitigation of colitis-associated tumorigenesis in Apc(Min/+) mice. J. Pharmacol. Exp. Ther. 2014, 350, 99-109.
6. Bu Y, Li X, He Y, Huang C, Shen Y, Cao Y, Huang D, Cai C, Wang Y, Wang Z, Liao DF, Cao D. A phosphomimetic mutant of RelA/p65 at Ser536 induces apoptosis and senescence: An implication for tumor-suppressive role of Ser536 phosphorylation. Int J Cancer. 2016; 138:1186-1198.
7. Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004; 25:280-288.
8. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006; 441:431-436.
9. Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat Immunol. 2011; 12:715-723.
10. Perkins ND. The diverse and complex roles of NF-κB subunits in cancer. Nat Rev Cancer. 2012; 12:121-132.
11. Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004; 118:285-296.
12. Guina T, Biasi F, Calfapietra S, Nano M, Poli G. Inflammatory and redox reactions in colorectal carcinogenesis. Ann N Y Acad Sci. 2015; 1340:95-103.
13. Fukushima T, Kawaguchi M, Yorita K, Tanaka H, Takeshima H, Umezawa K, Kataoka H. Antitumor effect of dehydroxymethylepoxyquinomicin, a small molecule inhibitor of nuclear factor-κB, on glioblastoma. Neuro Oncol. 2012; 14:19-28.
14. Horiuchi K, Morioka H, Nishimoto K, Suzuki Y, Susa M, Nakayama R, Kawai A, Sonobe H, Takaishi H, Ozaki T, Yabe H, Umezawa K, Toyama Y. Growth suppression and apoptosis induction in synovial sarcoma cell lines by a novel NF-kappaB inhibitor, dehydroxymethylepoxyquinomicin (DHMEQ). Cancer Lett. 2008; 272:336-344.
15. Meng Z, Mitsutake N, Nakashima M, Starenki D, Matsuse M, Takakura S, Namba H, Saenko V, Umezawa K, Ohtsuru A, Yamashita S. Dehydroxymethylepoxyquinomicin, a novel nuclear Factor-kappaB inhibitor, enhances antitumor activity of taxanes in anaplastic thyroid cancer cells. Endocrinology. 2008; 149:5357-5365.
16. Lampiasi N, Azzolina A, D’Alessandro N, Umezawa K, McCubrey JA, Montalto G, Cervello M. Antitumor effects of dehydroxymethylepoxyquinomicin, a novel nuclear factor-kappaB inhibitor, in human liver cancer cells are mediated through a reactive oxygen species-dependent mechanism. Mol Pharmacol. 2009; 76:290-300.
17. Kodaira K, Kikuchi E, Kosugi M, Horiguchi Y, Matsumoto K, Kanai K, Suzuki E, Miyajima A, Nakagawa K, Tachibana M, Umezawa K, Oya M. Potent cytotoxic effect of a novel nuclear factor-kappaB inhibitor dehydroxymethylepoxyquinomicin on human bladder cancer cells producing various cytokines. Urology. 2010; 75:805-812.
18. Mino K, Ozaki M, Nakanishi K, Haga S, Sato M, Kina M, Takahashi M, Takahashi N, Kataoka A, Yanagihara K, Ochiya T, Kamiyama T, Umezawa K, Todo S. Inhibition of nuclear factor-κB suppresses peritoneal dissemination of gastric cancer by blocking cancer cell adhesion. Cancer Sci. 2011; 102:1052-1058.
19. Yasuda A, Kondo S, Nagumo T, Tsukamoto H, Mukudai Y, Umezawa K, Shintani S. Anti-tumor activity of dehydroxymethylepoxyquinomicin against human oral squamous cell carcinoma cell lines in vitro and in vivo. Oral Oncol. 2011; 47:334-339.
20. Umezawa K. Possible role of peritoneal NF-κB in peripheral inflammation and cancer: lessons from the inhibitor DHMEQ. Biomed Pharmacother. 2011; 65:252-259.
21. Lampiasi N, Azzolina A, Umezawa K, Montalto G, McCubrey JA, Cervello M. The novel NF-κB inhibitor DHMEQ synergizes with celecoxib to exert antitumor effects on human liver cancer cells by a ROS-dependent mechanism. Cancer Lett. 2012; 322:35-44.
22. Castro-Gamero AM, Borges KS, da Silva Silveira V, Lira RCP, de Paula Gomes Queiroz R, Valera FCP, Scrideli CA, Umezawa K, Tone LG. Inhibition of nuclear factor-κB by dehydroxymethylepoxyquinomicin induces schedule-dependent chemosensitivity to anticancer drugs and enhances chemoinduced apoptosis in osteosarcoma cells. Anticancer Drugs. 2012; 23:638-650.
23. Kozakai N, Kikuchi E, Hasegawa M, Suzuki E, Ide H, Miyajima A, Horiguchi Y, Nakashima J, Umezawa K, Shigematsu N, Oya M. Enhancement of radiosensitivity by a unique novel NF-κB inhibitor, DHMEQ, in prostate cancer. Br J Cancer. 2012; 107:652-657.
24. Sato M, Nakanishi K, Haga S, Fujiyoshi M, Baba M, Mino K, Niwa H, Yokoo H, Umezawa K, Ohmiya Y, Kamiyama T, Todo S, Taketomi A, Ozaki M. Anoikis induction and inhibition of peritoneal metastasis of pancreatic cancer cells by a nuclear factor-κB inhibitor, (-)-DHMEQ. Oncol Res. 2013; 21:333-343.
25. Togano T, Nakashima M, Watanabe M, Umezawa K, Watanabe T, Higashihara M, Horie R. Synergistic effect of 5-azacytidine and NF-κB inhibitor DHMEQ on apoptosis induction in myeloid leukemia cells. Oncol Res. 2012; 20:571-577.
26. Togano T, Watanabe M, Itoh K, Umezawa K, Masuda N, Higashihara M, Horie R. Activation of Akt involves resistance to NF-κB inhibition and abrogation of both triggers synergistic apoptosis in lung adenocarcinoma cells. Lung Cancer. 2014; 83:139-145.
27. Kataoka H, Suganuma T, Shimomura T, Itoh H, Kitamura N, Nabeshima K, Koono M. Distribution of hepatocyte growth factor activator inhibitor type 1 (HAI-1) in human tissues. Cellular surface localization of HAI-1 in simple columnar epithelium and its modulated expression in injured and regenerative tissues. J Histochem Cytochem. 1999; 47:673-682.
28. Kawaguchi M, Takeda N, Hoshiko S, Yorita K, Baba T, Sawaguchi A, Nezu Y, Yoshikawa T, Fukushima T, Kataoka H. Membrane-bound serine protease inhibitor HAI-1 is required for maintenance of intestinal epithelial integrity. Am J Pathol. 2011; 179:1815-1826.
29. Hoshiko S, Kawaguchi M, Fukushima T, Haruyama Y, Yorita K, Tanaka H, Seiki M, Inatsu H, Kitamura K, Kataoka H. Hepatocyte growth factor activator inhibitor type 1 is a suppressor of intestinal tumorigenesis. Cancer Res. 2013; 73:2659-2670.
30. Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990; 247:322-324.
31. Kawaguchi M, Kataoka H. Mechanisms of Hepatocyte Growth Factor Activation in Cancer Tissues. Cancers (Basel). 2014; 6:1890-1904.
32. Hao NB, Tang B, Wang GZ, Xie R, Hu CJ, Wang SM, Wu YY, Liu E, Xie X, Yang SM. Hepatocyte growth factor (HGF) upregulates heparanase expression via the PI3K/Akt/NF-κB signaling pathway for gastric cancer metastasis. Cancer Lett. 2015; 361:57-66.
33. Esencay M, Newcomb EW, Zagzag D. HGF upregulates CXCR4 expression in gliomas via NF-kappaB: implications for glioma cell migration. J Neurooncol. 2010; 99:33-40.
34. Nalla AK, Gogineni VR, Gupta R, Dinh DH, Rao JS. Suppression of uPA and uPAR blocks radiation-induced MCP-1 mediated recruitment of endothelial cells in meningioma. Cell Signal. 2011; 23:1299-1310.
35. Abraham E, Gyetko MR, Kuhn K, Arcaroli J, Strassheim D, Park JS, Shetty S, Idell S. Urokinase-type plasminogen activator potentiates lipopolysaccharide-induced neutrophil activation. J Immunol. 2003; 170:5644-5651.
36. Mori Y, Akita K, Tanida S, Ishida A, Toda M, Inoue M, Yashiro M, Sawada T, Hirakawa K, Nakada H. MUC1 protein induces urokinase-type plasminogen activator (uPA) by forming a complex with NF-κB p65 transcription factor and binding to the uPA promoter, leading to enhanced invasiveness of cancer cells. J Biol Chem. 2014; 289:35193-35204.
37. Moran P, Li W, Fan B, Vij R, Eigenbrot C, Kirchhofer D. Pro-urokinase-type plasminogen activator is a substrate for hepsin. J Biol Chem. 2006; 281:30439-30446.
38. Wojtukiewicz MZ, Hempel D, Sierko E, Tucker SC, Honn KV. Protease-activated receptors (PARs)-biology and role in cancer invasion and metastasis. Cancer Metastasis Rev. 2015; 34:775-796.
39. Kawaguchi M, Kanemaru A, Sawaguchi A, Yamamoto K, Baba T, Lin CY, Johnson MD, Fukushima T, Kataoka H. Hepatocyte growth factor activator inhibitor type 1 maintains the assembly of keratin into desmosomes in keratinocytes by regulating protease-activated receptor 2-dependent p38 signaling. Am J Pathol. 2015; 185:1610-1623.
40. Scott G, Leopardi S, Parker L, Babiarz L, Seiberg M, Han R. The proteinase-activated receptor-2 mediates phagocytosis in a Rho-dependent manner in human keratinocytes. J Invest Dermatol. 2003; 121:529-541.
41. Driesbaugh KH, Buzza MS, Martin EW, Conway GD, Kao JPY, Antalis TM. Proteolytic activation of the protease-activated receptor (PAR)-2 by the glycosylphosphatidylinositol-anchored serine protease testisin. J Biol Chem. 2015; 290:3529-3541.
42. Johnson JJ, Miller DL, Jiang R, Liu Y, Shi Z, Tarwater L, Williams R, Balsara R, Sauter ER, Stack MS. Protease Activated Receptor-2 (PAR-2)-Mediated Nf-κB Activation Suppresses Inflammation-Associated Tumor Suppressor MicroRNAs in Oral Squamous Cell Carcinoma. J Biol Chem. 2016; 291:6936-6946.
43. Sales KU, Friis S, Konkel JE, Godiksen S, Hatakeyama M, Hansen KK, Rogatto SR, Szabo R, Vogel LK, Chen W, Gutkind JS, Bugge TH. Non-hematopoietic PAR-2 is essential for matriptase-driven pre-malignant progression and potentiation of ras-mediated squamous cell carcinogenesis. Oncogene. 2014; 34:346-356.