
Introduction
EMT and signaling
EMT is an ancient developmental process that is characterized by morphological changes in epithelial cells, whereby they acquire a mesenchyme phenotype [1]. The hallmarks of EMT are functionally decreased adhesive capacity and increased mobility [2]. This is facilitated by epithelial phenotype changes where cells switch from apical-basal polarity to anterior-posterior polarity, characteristically illustrated with disrupted intercellular junctions and increased migratory potential of individual cells. The basis of EMT in physiology is to remove/replace unnecessary epithelia at specific locations or developmental stages. Biochemically, EMT cells exhibit distinct patterns of upregulated gene expression of proteins engaged in remodeling of cell-cell contact, cytoskeleton, and interaction with extracellular matrix (ECM) [3]. In the last two decades, a group of transcription factors including the Twist, Snail and Zeb families, have been identified as the EMT-inducing transcription factors governing the EMT process in vitro, as inducers, enhancers, or both [4]. Overexpression of these proteins in untransformed mammalian epithelial cells induces potent cell scattering phenotypes that also functionally resemble the original EMT in mouse model [5, 6]. Therefore, the EMT process is generally recognized as a reprogramming event essentially driven at the transcriptional level, whilst being initiated and coupled with ECM signaling [7, 8] (Figure 1).
Many pathways are associated with EMT. One of the well-documented signaling axes is the transforming growth factor (TGF) cascade [9] that induces cell scattering with increased invasive potential in a variety of cancer cells with epithelial origin [10, 11]. In addition to TGFβ, the receptor tyrosine kinases (RTKs) such as EGFR family [12], c-Met [13], VEGFR [14, 15], PDGFR [16] and others (such as Wnt pathway [17, 18]) are all capable of initiating and/or maintaining EMT phenotypes. Importantly, activation of these membrane-associated signaling complexes often correlates with elevated expression level of the EMT-inducing transcription factors, and conversely, in addition to maintaining the EMT gene signature, can also maintain and enhance RTK signaling through feedback or feed-forward signaling loops [19, 20]. With a particular focus on understanding these mechanisms connecting the EMT transcriptional response with the activation of the upstream signalosomes, a number of studies revealed that three intracellular cascades, mTOR/PI3K/Akt, MAPK and Rho GTPases are core mediators transducing signals to activate the EMT-inducing transcription factors [21-24]. These findings have been demonstrated in many types of carcinomas, with great consistency [25].
The invasive behaviors of metastatic cancer cells closely resemble the physiological EMT phenotypes and effectively respond to the known upstream signaling cascades. Metastasis initiates with a small portion of the cancer cells within the primary organ disseminating from the tumor mass and by expressing remarkably high level of proteases, digest matrix barriers and subsequently invade into surrounding tissues [26, 27]. Local invasive cancer cells further intravasate into lymphatic and/or blood vessels and transit to distant organs where they extravasate and re-colonize as a metastatic tumour lesion [28, 29]. A specific biological impact of the EMT inducers in cancer metastasis is demonstrated by depletion of the EMT inducers in invasive cancer cells, which significantly attenuates the metastatic spread, whilst in most cases having little to no effect on the primary tumor growth [30]. This indicates that cancer cells preferentially take advantage of the physiological EMT signaling to support their invasion.
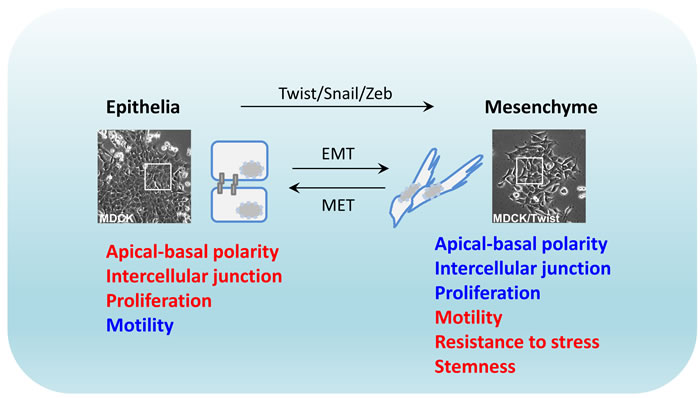
Figure 1: Plastic epithelial-to-mesenchymal transition. Transcriptional regulation of EMT by activated Twist, Snail and Zeb oncogenic proteins is often accompanied with cellular morphological change. EMT: epithelial-mesenchymal transition; MET: mesenchymal-epithelial transition. Red: high level; blue: low level.
The plasticity of EMT
Although TGF signaling triggers robust EMT activation in epithelial cells, a reversal of the EMT phenotype was observed when TGFβ signaling was disrupted by withdrawal [31] or pharmacological inhibition [32], indicating a high degree of plasticity in the EMT process [33]. In an in vivo orthotopic metastatic mouse model, such reversal was discovered in the cancer cells that had undergone EMT to colonize and establish the metastatic tumour. It was found that the activity of Twist was significantly reduced at the initial phase of metastatic colonization in the lung and the cancer cells re-exhibited a classical epithelial phenotype with decreased migratory potential, with this process being coined MET (mesenchymal-to-epithelial transition) [34]. In contrast to the EMT phenotype which supports the cancer cell invasion but restricts their proliferation, MET favors re-activation of the proliferative potential, but limits invasiveness. In addition to Twist, other EMT inducers also displayed downregulated activity during the establishment of metastatic growth [35], and MET was postulated to be essential to maintain cancer cell survival in metastatic sites [36, 37]. Moreover, experimental evidence also demonstrated that the cells with phenotypic characters of EMT antagonize stress-induced apoptosis, including resistance to hypoxic pressure and DNA-damaging reagents. These observations support a hypothesis that the cancer cells with EMT properties potentially possess a “stemness” capacity [38-40], although many aspects of this concept are still to be convincingly demonstrated [41].
Regulation of the EMT-inducing transcription factor Twist
Considering the interplay between membrane-associated signaling complexes and EMT-inducing transcription factors, as well as the impact of EMT on cancer metastasis, it is essential to understand the fundamental mechanisms of how signals are transduced to activate the EMT inducers such as Twist. Twist belongs to the basic helix-loop-helix (bHLH) transcription factor family. In mammals, Twist exists as two forms that are crucial for proper prenatal development [42, 43], although their functional roles in postnatal physiology are still vague. Across species from fruit fly to human, it is evolutionarily conserved and recognizes a palindromic-like consensus sequence CANNTG, also called E-box, in the proximal region of promoters. Its binding capacity is preferentially mediated through hetero-dimerization of Twist with other family members to implement its transcriptional regulation [44]. A recent study employing ChIP coupled with high-throughput sequencing for the analysis of Twist-binding DNA elements revealed that Twist can bind to two tandem E-boxes, a unique feature that distinguishes it from other bHLH transcription factors [45]. To date, clinical studies have shown evidence to support a pro-metastatic role for Twist regulated EMT gene expression during cancer progression [46-49]. This includes a number of studies indicating that overexpression of Twist enhances carcinoma metastasis [50, 51] and associates with unfavorable clinical prognosis [52-55]. Mechanistically, Twist-promoted cancer metastasis is mediated through its transcription activity that is hijacked by cancer cells [25]. Twist binding to E-boxes can transcriptionally repress E-cadherin expression, and consequently disrupts the intercellular adhesion and induce single cancer cell dissemination from the primary location [56]. In parallel, Twist overexpression remodels cytoskeleton and upregulates several essential signaling molecules such as Akt2 and TGFβ2 to robustly induce the EMT phenotype [57].
Differential phosphorylation of Twist as a functional switch
Interestingly, TGFβ2 upregulation by Twist can be enhanced by Akt-mediated phosphorylation of Twist on serine 42 (S42), which increases binding to the TGFβ2 promoter. Thus, Akt-directed Twist phosphorylation on S42 is crucial for the crosstalk between PI3K/Akt and TGFβ pathways in metastatic breast cancer [57] and is also demonstrated to be an invasive signature in other cancer models [58]. Not limited to S42 phosphorylation, independent studies have highlighted other phosphorylation sites on Twist including serine 68 (S68) [59], serine 18 (S18) and serine 20 (S20) [60], as well as threonine 121 (T121) and serine 123 (S123) [61] that are differentially phosphorylated by MAPK (JNK, ERK, p38), casein kinase 2 and Akt, respectively, most of which are suggested to activate and/or enhance Twist functions through promoting its stability in a context-dependent manner. Analysis of the protein sequences of Twist family members in mammals demonstrates that all these crucial phosphorylation sites are evolutionarily conserved (Figure 2), implying that the Twist proteins across species potentially share a functional homology and the two Twist isoforms may display redundant functions. Clearly, Twist-phosphorylating kinases are mainly the two important intracellular signaling mediators Akt and MAPK, both of which are indisputably involved in cell proliferation, differentiation and invasion in cancer cells and key players of drug resistance in clinic. These two nodes can be activated by drivers of EMT and metastasis including well-known receptor signaling kinases such as TGFβ, RTKs, ECM-mediated integrin pathway and canonical or non-canonical WNT signaling, which in turn facilitate EMT and cancer metastasis via regulation of Akt and MAPK. The increasing evidence demonstrating the activity of Twist in regulation of cell migration and invasion which are controlled by a posttranslational modifications may have many critical implications for controlling metastatic lesions, when cancer cells need to re-establish high levels of proliferation and growth. Intriguingly, the basal level of Twist in non-neoplastic or non-metastatic cells is generally low, indicating that Twist activation starts from an activated transcription program. To date, it has been shown that dependent upon the types of malignancy, Twist can be transcriptionally upregulated by NF-κB [62, 63], STAT3 [64-66], DLX4 [67], MYCN and MYC [68], HMGA2 [69] and SOX2 [70],, indicating that activation of Twist is a coordinated event between epigenetic transcriptional regulation and post-translational modification. Following phospho-activation, as a core element involved in the activation of transcriptional complexes, Twist can drive the transcription of a number of target genes, many of which are oncogenic. Although Twist was shown to be a transcriptional repressor that inversely correlates with E-cadherin expression [71], it may possibly not be directly involved in suppressing CDH1 transcription, rather, this is suggested to be mediated by Snail2, a direct transcription target downstream of Twist [72]. It should also be noted that the activation of Snail proteins, like Twist, is also under the control of phosphorylation [73]. Depending on the upstream kinases, phosphorylated Snail may exhibit enhanced repressing activity [74], or rapid degradation [75]. Direct binding of Snail proteins within the regulatory region of CDH1 has been reported to mediate its repressing [76] (Figure 3). Thus, it seems that the repression of CDH1 transcription is a sequential program controlled by phospho-activated Twist/Snail axis. A recent study focusing on the specificity of Twist-mediated transcription, revealed that Twist dimers can uniquely recognized a tandem stretch of E-boxes [45]. The biological consequence of such interaction in relation to preferential to transcriptional activation, repression, or both, remains to be determined. Undoubtedly, whether any related transcriptional specificity is correlated with the multifaceted phosphorylation patterns is also an essential question.
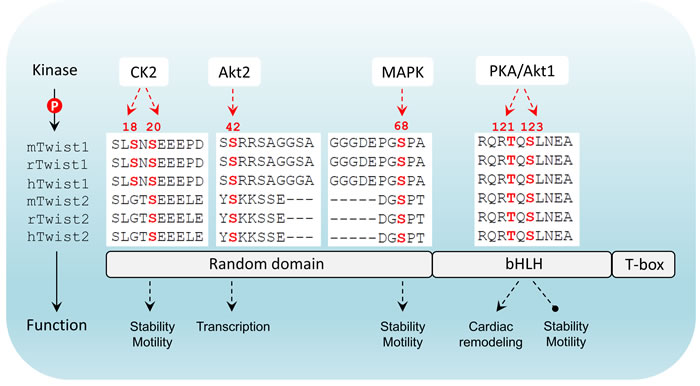
Figure 2: Twist structure and phosphorylation conservation in mammals. Twist has three major domains including a N-terminal flexible domain, a basic helix-loop-helix domain that is responsible for DNA-binding and a C-terminal Twist-box. Differential phosphorylation patterns that have been reported are indicated. In mammals there are two members in Twist family. The phosphorylated amino acids highlighted in red are highly conserved in both members across three species (m: mouse; r: rat; h: human). CK2: casein kinase 2; bHLH: basic helix-loop-helix; T-box: Twist-box.
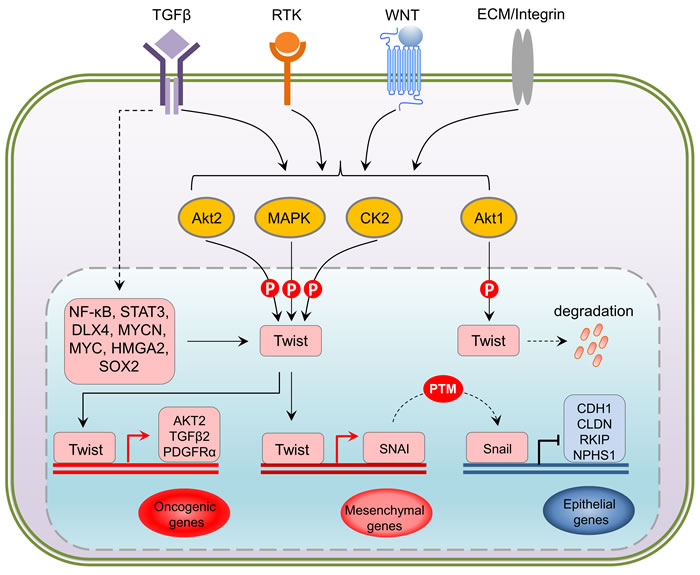
Figure 3: Upstream and downstream regulation of Twist. At transcriptional level, NF-κB, STAT3, DLX4, MYCN, MYC, HMGA2 and SOX2 have been shown to upregulate Twist in response to the activation of TGFbeta, RTKs, WNT and Integrin pathways. Being a central hub, translated Twist undergoes differential phosphorylation directly mediated by Akt, MAPK and CK2 kinases in a context-dependent manner. Activation of Twist via phosphorylation triggers oncogenic gene expression such as AKT, TGFB and PDGFR, and represses E-cadherin (CDH1), Claudins (CLDN), Raf kinase inhibitor protein (RKIP) and Nephrin1 (NPHS1) that are crucial for cell-cell contact through its downstream target Snail, an epigenetic event that actively leads to an reinfoced EMT activation. PTM: post-translational modification.
Inhibition and reversal of Twist-induced EMT at the site of metastasis
The metastasizing cancer cells displaying an EMT phenotype are found to exhibit higher resistance to environmental stress [77, 78]. High expression level of EMT signatures observed in circulating invasive cells supports survival in the blood stream [79, 80] , allowing subsequent extravasation of these cells from the blood or lymphatic vessels and establishment in distant organs/tissues. Suppression of the EMT capability in the circulating cancer cells can efficiently attenuate the anti-apoptotic competence [81]. Therefore, the EMT state actively supports cancer cells to overcome environmental stress. As mentioned above, the signaling mediators that link extracellular signals to EMT inducers often converge to two major signaling nodes PI3K/Akt and MAPK, both of which are responsible for drug resistance to clinical therapies in many types of cancer [82]. Indeed, the metastatic cancer cells associated with EMT clearly show remarkable resistance to a number of small molecular inhibitors in clinic [83-85]. As EMT programming limits cell proliferation, it is critical for re-establishing cancer cells at the distant metastatic site to overcome this barrier for colonization. In this regard, it raises the fundamental question of how EMT is revered, or more specifically how EMT inducers, such as Twist, are deactivated. Whilst further insights are being made, the current understanding remains somewhat limited, with changes in Twist stability the best described. In different types of cancer cells, Twist instability can be induced by changes in microRNA [86-88]. In addition to the regulation of Twist stability at RNA level, two notable studies suggest that deactivation of Twist in metastatic lesion may be triggered by elevated protein instability, mediated by either de-phosphorylation of S68 by the small C-terminal Domain Phosphatase 1 [89], or Akt1-directed phosphorylation of Twist on T121 and S123 that promotes β-TrCP-mediated Twist1 ubiquitination and degradation [90]. These two discoveries elucidate the suppression of Twist1 activity through potential kinase-regulated autonomous feedback signaling and further highlights the developing contribution of phosphorylation to the regulation of the EMT process.
Discussion
Metastatic spread is a key issue in clinical cancer therapy because it represents the major cause of death for cancer patients. Many types of invasive cancer with epithelial origin represent an advanced EMT phenotype that exhibits a high grade of plasticity. Establishment and outgrowth of metastases in distant organs primarily requires local re-colonization, which is reflected by decreased motility and increased proliferation potential of the cancer cells. Such reversal from EMT to MET in breast cancer-associated metastases was experimentally recognized in Twist-induced metastatic models [34, 35]. However, the mechanisms of how Twist activity is regulated during the establishment of metastases is currently undetermined. What factors in the metastatic site determine the need of its inactivation? Recent studies in mouse model shed some lights on these issues [90]. Cancer cell proliferation in the metastatic site requires high level of global Akt activity which is principally capable of inducing cell migration, and Twist was reported to inhibit environmental stress-triggered senescence [91, 92]. The opposing roles of Akt1 and Akt2 during metastatic progression [93, 94] may provide a ration interpretation by proposing a model whereby when cancer cells need to disseminate, Akt2-mediated phosphorylation on S42 of Twist is dominant; whilst during metastatic colonization, Akt1-mediated phosphorylation on T121 and S123 triggers ubiquitination-dependent degradation of Twist and promotes proliferation. This would imply the existence of a dynamic pool of Akt isoforms that differentially regulates cancer cell behavior at both primary and metastatic sites. If this is true in vivo, the predominance of individual Akt isoforms will be the fundamental factor to determining the cancer cell behavior and fate. Interestingly, independent study has shown that Akt isoforms are individually regulated by specific microRNAs thus distinguishes the spatiotemporal Akt isoforms-contributed activity [95].
This model whereby Twists oncogenic functions are regulated by the predominant Akt isoforms raises further crucial questions to be elucidated : 1) what are the signals in cancer cells that would control the switch of Twist-phosphorylating modulators between the Akt1 and Akt2 isoforms? 2) How do Akt1 and Akt2 compete with each other to impose dominant control of Twist phosphorylation? 3) What controls the Akt1 and Akt2 ratio specifically in the metastatic lesion? These questions still remain unanswered and studies have reported contradictory observations, including a structural study of Twist linking T121 and S123 phosphorylation in prostate cancer to Twist mediated metastasis, as opposed to Twist degradation [96]. This discrepancy may indicate difference between cancer types or progression stages. An explanation for the dominating status of Akt1 and Akt2 may be that there exists a feed-back signaling loop through multiple signaling cross-talks that eventually leads to microRNA-mediated degradation of individual Akt isoforms, thereby modulating entire Akt pool activity [95, 97]. Alternatively, it is also proposed that distinct phosphorylation patterns of Twist require different co-activators, which may determine the selectivity of phosphorylation motifs on Twist exposed to Akt isoforms. In fact, such phospho-specific phenotypes coupled with distinct cell fate in cancer have been also found similar to other EMT-drivers such as Snail family [75, 98]. Thus, phosphorylation of EMT-promoting molecules seems to, at least in part, elaborately act as a functional “on/off” signal in favor of cancer cell migration, invasion and survival. Moreover, on-site de-phosphorylation by specific phosphatases could also contribute, either directly through targeting Twist, or indirectly by deactivating upstream kinases such as tyrosine receptors, to influence Twist stability. Insights into this may be found in the activity of SHP2 in a well-studied metastasis model, which is tremendously increased and promotes metastatic colonization [99].
Additionally, more experimental data is needed to explore the mechanistic interplay between Twist (and/or others)-induced EMT and metastasis. A recent study challenged the conventional role of Twist-induced EMT in promotion of metastasis. In a pancreatic cancer model, depletion of Twist or Snail did not inhibit pancreatic metastasis; rather, EMT was crucial for the tumors to resist to the treatment of DNA-damaging reagents [100], another important notion closely related to current clinical cancer therapies [101, 102]. Moreover, it would be interesting to know whether Twist phosphorylation on different sites occurs simultaneously, individually or step-wisely. Does one site phosphorylation/de-phosphorylation influence the others? The answers to these questions will facilitate a better understanding of the affiliated signaling events that regulate the transition between EMT and MET during cancer progression, which will greatly facilitate development of diagnostic tools for clinical applications. Furthermore, such phospho-pattern-specific biomarkers may not only predict whether the cancer cells in metastatic tumors start a second wave of dissemination, but also be considered as potential druggable targets [103].
Acknowledgments
We thank Dr. David Restuccia for the critical comments and discussions. This work was supported by National Natural Science Foundation of China Grant 381570056 (to H. Tang), Fondazione Cassa di Risparmio di Pistoia&Pescia (ID 154/2014) (to D. Massi), Associazione Italiana per la Ricerca sul Cancro (A.I.R.C. 5xmille Ref. 12237) P.I. A.F. (to M. Mandalà), Swiss Cancer Res Foundation Grants KFS-3170-02-2013 and KFS-3501-08-2014 (to A. Wicki), and Swiss National Science Foundation Grant 31-130838 (to B. A. Hemmings and G. Xue).
Conflicts of Interest
The authors declare no potential conflicts of interest.
References
1. Hay ED. An overview of epithelio-mesenchymal transformation. Acta anatomica. 1995; 154(1):8-20.
2. Kalluri R and Weinberg RA. The basics of epithelial-mesenchymal transition. The Journal of clinical investigation. 2009; 119(6):1420-1428.
3. Polyak K and Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nature reviews Cancer. 2009; 9(4):265-273.
4. Sanchez-Tillo E, Liu Y, de Barrios O, Siles L, Fanlo L, Cuatrecasas M, Darling DS, Dean DC, Castells A and Postigo A. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. CMLS. 2012; 69(20):3429-3456.
5. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, Leach SD and Stanger BZ. EMT and dissemination precede pancreatic tumor formation. Cell. 2012; 148(1-2):349-361.
6. Cardiff RD. The pathology of EMT in mouse mammary tumorigenesis. Journal of mammary gland biology and neoplasia. 2010; 15(2):225-233.
7. Wu CY, Tsai YP, Wu MZ, Teng SC and Wu KJ. Epigenetic reprogramming and post-transcriptional regulation during the epithelial-mesenchymal transition. TIG. 2012; 28(9):454-463.
8. Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. The Journal of clinical investigation. 2009; 119(6):1417-1419.
9. Xu J, Lamouille S and Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell research. 2009; 19(2):156-172.
10. Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, Downward J, Beug H and Grunert S. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. The Journal of cell biology. 2002; 156(2):299-313.
11. Zavadil J and Bottinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005; 24(37):5764-5774.
12. Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei Y, Abbruzzese JL, Hortobagyi GN and Hung MC. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer research. 2007; 67(19):9066-9076.
13. Toiyama Y, Yasuda H, Saigusa S, Matushita K, Fujikawa H, Tanaka K, Mohri Y, Inoue Y, Goel A and Kusunoki M. Co-expression of hepatocyte growth factor and c-Met predicts peritoneal dissemination established by autocrine hepatocyte growth factor/c-Met signaling in gastric cancer. International journal of cancer. 2012; 130(12):2912-2921.
14. Fantozzi A, Gruber DC, Pisarsky L, Heck C, Kunita A, Yilmaz M, Meyer-Schaller N, Cornille K, Hopfer U, Bentires-Alj M and Christofori G. VEGF-mediated angiogenesis links EMT-induced cancer stemness to tumor initiation. Cancer research. 2014; 74(5):1566-1575.
15. Gonzalez-Moreno O, Lecanda J, Green JE, Segura V, Catena R, Serrano D and Calvo A. VEGF elicits epithelial-mesenchymal transition (EMT) in prostate intraepithelial neoplasia (PIN)-like cells via an autocrine loop. Experimental cell research. 2010; 316(4):554-567.
16. Jechlinger M, Sommer A, Moriggl R, Seither P, Kraut N, Capodiecci P, Donovan M, Cordon-Cardo C, Beug H and Grunert S. Autocrine PDGFR signaling promotes mammary cancer metastasis. The Journal of clinical investigation. 2006; 116(6):1561-1570.
17. Chang YW, Su YJ, Hsiao M, Wei KC, Lin WH, Liang CL, Chen SC and Lee JL. Diverse Targets of beta-Catenin during the Epithelial-Mesenchymal Transition Define Cancer Stem Cells and Predict Disease Relapse. Cancer research. 2015; 75(16):3398-3410.
18. Xue G, Romano E, Massi D and Mandala M. Wnt/beta-catenin signaling in melanoma: Preclinical rationale and novel therapeutic insights. Cancer treatment reviews. 2016; 49:1-12.
19. Sosic D, Richardson JA, Yu K, Ornitz DM and Olson EN. Twist regulates cytokine gene expression through a negative feedback loop that represses NF-kappaB activity. Cell. 2003; 112(2):169-180.
20. Eckert MA and Yang J. Targeting invadopodia to block breast cancer metastasis. Oncotarget. 2011; 2(7):562-568. doi: 10.18632/oncotarget.301
21. Larue L and Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3’ kinase/AKT pathways. Oncogene. 2005; 24(50):7443-7454.
22. Siegel PM and Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nature reviews Cancer. 2003; 3(11):807-821.
23. Mulholland DJ, Kobayashi N, Ruscetti M, Zhi A, Tran LM, Huang J, Gleave M and Wu H. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer research. 2012; 72(7):1878-1889.
24. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nature reviews Cancer. 2002; 2(6):442-454.
25. De Craene B and Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nature reviews Cancer. 2013; 13(2):97-110.
26. Keirsse J, Laoui D, Van Overmeire E and Van Ginderachter JA. Targeting cell-intrinsic and cell-extrinsic mechanisms of intravasation in invasive breast cancer. Science signaling. 2014; 7(353):pe28.
27. Kim J, Yu W, Kovalski K and Ossowski L. Requirement for specific proteases in cancer cell intravasation as revealed by a novel semiquantitative PCR-based assay. Cell. 1998; 94(3):353-362.
28. Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF and Groom AC. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. The American journal of pathology. 1998; 153(3):865-873.
29. Nguyen DX, Bos PD and Massague J. Metastasis: from dissemination to organ-specific colonization. Nature reviews Cancer. 2009; 9(4):274-284.
30. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A and Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004; 117(7):927-939.
31. Muraoka RS, Dumont N, Ritter CA, Dugger TC, Brantley DM, Chen J, Easterly E, Roebuck LR, Ryan S, Gotwals PJ, Koteliansky V and Arteaga CL. Blockade of TGF-beta inhibits mammary tumor cell viability, migration, and metastases. The Journal of clinical investigation. 2002; 109(12):1551-1559.
32. Gal A, Sjoblom T, Fedorova L, Imreh S, Beug H and Moustakas A. Sustained TGF beta exposure suppresses Smad and non-Smad signalling in mammary epithelial cells, leading to EMT and inhibition of growth arrest and apoptosis. Oncogene. 2008; 27(9):1218-1230.
33. Nieto MA and Cano A. The epithelial-mesenchymal transition under control: global programs to regulate epithelial plasticity. Seminars in cancer biology. 2012; 22(5-6):361-368.
34. Tsai JH, Donaher JL, Murphy DA, Chau S and Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer cell. 2012; 22(6):725-736.
35. Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, Barrallo-Gimeno A, Cano A and Nieto MA. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer cell. 2012; 22(6):709-724.
36. Chaffer CL, Brennan JP, Slavin JL, Blick T, Thompson EW and Williams ED. Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer research. 2006; 66(23):11271-11278.
37. Korpal M, Ell BJ, Buffa FM, Ibrahim T, Blanco MA, Celia-Terrassa T, Mercatali L, Khan Z, Goodarzi H, Hua Y, Wei Y, Hu G, Garcia BA, et al. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nature medicine. 2011; 17(9):1101-1108.
38. Medema JP. Cancer stem cells: the challenges ahead. Nature cell biology. 2013; 15(4):338-344.
39. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133(4):704-715.
40. Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S and Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PloS one. 2008; 3(8):e2888.
41. Beck B, Lapouge G, Rorive S, Drogat B, Desaedelaere K, Delafaille S, Dubois C, Salmon I, Willekens K, Marine JC and Blanpain C. Different levels of Twist1 regulate skin tumor initiation, stemness, and progression. Cell stem cell. 2015; 16(1):67-79.
42. Kang Y and Massague J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell. 2004; 118(3):277-279.
43. Rice DP, Aberg T, Chan Y, Tang Z, Kettunen PJ, Pakarinen L, Maxson RE and Thesleff I. Integration of FGF and TWIST in calvarial bone and suture development. Development. 2000; 127(9):1845-1855.
44. Castanon I, Von Stetina S, Kass J and Baylies MK. Dimerization partners determine the activity of the Twist bHLH protein during Drosophila mesoderm development. Development. 2001; 128(16):3145-3159.
45. Chang AT, Liu Y, Ayyanathan K, Benner C, Jiang Y, Prokop JW, Paz H, Wang D, Li HR, Fu XD, Rauscher FJ, 3rd and Yang J. An evolutionarily conserved DNA architecture determines target specificity of the TWIST family bHLH transcription factors. Genes & development. 2015; 29(6):603-616.
46. Lee TK, Poon RT, Yuen AP, Ling MT, Kwok WK, Wang XH, Wong YC, Guan XY, Man K, Chau KL and Fan ST. Twist overexpression correlates with hepatocellular carcinoma metastasis through induction of epithelial-mesenchymal transition. Clin cancer res. 2006; 12(18):5369-5376.
47. Sossey-Alaoui K, Pluskota E, Davuluri G, Bialkowska K, Das M, Szpak D, Lindner DJ, Downs-Kelly E, Thompson CL and Plow EF. Kindlin-3 enhances breast cancer progression and metastasis by activating Twist-mediated angiogenesis. FASEB journal. 2014; 28(5):2260-2271.
48. Puisieux A, Valsesia-Wittmann S and Ansieau S. A twist for survival and cancer progression. British journal of cancer. 2006; 94(1):13-17.
49. Thiery JP and Morgan M. Breast cancer progression with a Twist. Nature medicine. 2004; 10(8):777-778.
50. Gajula RP, Chettiar ST, Williams RD, Thiyagarajan S, Kato Y, Aziz K, Wang R, Gandhi N, Wild AT, Vesuna F, Ma J, Salih T, Cades J, et al. The twist box domain is required for Twist1-induced prostate cancer metastasis. MCR. 2013; 11(11):1387-1400.
51. Shiota M, Yokomizo A, Tada Y, Inokuchi J, Kashiwagi E, Masubuchi D, Eto M, Uchiumi T and Naito S. Castration resistance of prostate cancer cells caused by castration-induced oxidative stress through Twist1 and androgen receptor overexpression. Oncogene. 2010; 29(2):237-250.
52. Shibata K, Kajiyama H, Ino K, Terauchi M, Yamamoto E, Nawa A, Nomura S and Kikkawa F. Twist expression in patients with cervical cancer is associated with poor disease outcome. Annals of oncology. 2008; 19(1):81-85.
53. Kajiyama H, Shibata K, Umezu T, Mizuno M, Suzuki S, Yamamoto E, Fujiwara S and Kikkawa F. Expression of Twist enhances risk of poor oncologic outcome in patients with stage Ib to II cervical carcinoma with lymphovascular space involvement. Human pathology. 2013; 44(2):181-188.
54. Nordfors K, Haapasalo J, Makela K, Granberg KJ, Nykter M, Korja M, Paavonen T, Haapasalo H and Soini Y. Twist predicts poor outcome of patients with astrocytic glioma. Journal of clinical pathology. 2015; 68(11):905-912.
55. Pereira L, Horta S, Mateus R and Videira MA. Implications of Akt2/Twist crosstalk on breast cancer metastatic outcome. Drug discovery today. 2015; 20(9):1152-1158.
56. Onder TT, Gupta PB, Mani SA, Yang J, Lander ES and Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer research. 2008; 68(10):3645-3654.
57. Xue G, Restuccia DF, Lan Q, Hynx D, Dirnhofer S, Hess D, Ruegg C and Hemmings BA. Akt/PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-beta signaling axes. Cancer discovery. 2012; 2(3):248-259.
58. Mammoto T, Jiang A, Jiang E and Mammoto A. The Role of Twist1 Phosphorylation in Angiogenesis and Pulmonary Fibrosis. American journal of respiratory cell and molecular biology. 2016.
59. Hong J, Zhou J, Fu J, He T, Qin J, Wang L, Liao L and Xu J. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer research. 2011; 71(11):3980-3990.
60. Su YW, Xie TX, Sano D and Myers JN. IL-6 stabilizes Twist and enhances tumor cell motility in head and neck cancer cells through activation of casein kinase 2. PloS one. 2011; 6(4):e19412.
61. Lu S, Nie J, Luan Q, Feng Q, Xiao Q, Chang Z, Shan C, Hess D, Hemmings BA and Yang Z. Phosphorylation of the Twist1-family basic helix-loop-helix transcription factors is involved in pathological cardiac remodeling. PloS one. 2011; 6(4):e19251.
62. Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, Chao CH, Yamaguchi H, Yang NK, Ding Q, Wang Y, Lai YJ, LaBaff AM, et al. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer research. 2012; 72(5):1290-1300.
63. Pham CG, Bubici C, Zazzeroni F, Knabb JR, Papa S, Kuntzen C and Franzoso G. Upregulation of Twist-1 by NF-kappaB blocks cytotoxicity induced by chemotherapeutic drugs. Molecular and cellular biology. 2007; 27(11):3920-3935.
64. Cheng GZ, Zhang WZ, Sun M, Wang Q, Coppola D, Mansour M, Xu LM, Costanzo C, Cheng JQ and Wang LH. Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. The Journal of biological chemistry. 2008; 283(21):14665-14673.
65. Hsu KW, Hsieh RH, Huang KH, Fen-Yau Li A, Chi CW, Wang TY, Tseng MJ, Wu KJ and Yeh TS. Activation of the Notch1/STAT3/Twist signaling axis promotes gastric cancer progression. Carcinogenesis. 2012; 33(8):1459-1467.
66. Cho KH, Jeong KJ, Shin SC, Kang J, Park CG and Lee HY. STAT3 mediates TGF-beta1-induced TWIST1 expression and prostate cancer invasion. Cancer letters. 2013; 336(1):167-173.
67. Zhang L, Yang M, Gan L, He T, Xiao X, Stewart MD, Liu X, Yang L, Zhang T, Zhao Y and Fu J. DLX4 upregulates TWIST and enhances tumor migration, invasion and metastasis. International journal of biological sciences. 2012; 8(8):1178-1187.
68. Selmi A, de Saint-Jean M, Jallas AC, Garin E, Hogarty MD, Benard J, Puisieux A, Marabelle A and Valsesia-Wittmann S. TWIST1 is a direct transcriptional target of MYCN and MYC in neuroblastoma. Cancer letters. 2015; 357(1):412-418.
69. Thuault S, Valcourt U, Petersen M, Manfioletti G, Heldin CH and Moustakas A. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. The Journal of cell biology. 2006; 174(2):175-183.
70. Bisaro B, Montani M, Konstantinidou G, Marchini C, Pietrella L, Iezzi M, Galie M, Orso F, Camporeale A, Colombo SM, Di Stefano P, Tornillo G, Camacho-Leal MP, et al. p130Cas/Cyclooxygenase-2 axis in the control of mesenchymal plasticity of breast cancer cells. BCR. 2012; 14(5):R137.
71. Vesuna F, van Diest P, Chen JH and Raman V. Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochemical and biophysical research communications. 2008; 367(2):235-241.
72. Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ and Yang J. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer research. 2011; 71(1):245-254.
73. Dominguez D, Montserrat-Sentis B, Virgos-Soler A, Guaita S, Grueso J, Porta M, Puig I, Baulida J, Franci C and Garcia de Herreros A. Phosphorylation regulates the subcellular location and activity of the snail transcriptional repressor. Molecular and cellular biology. 2003; 23(14):5078-5089.
74. Yang Z, Rayala S, Nguyen D, Vadlamudi RK, Chen S and Kumar R. Pak1 phosphorylation of snail, a master regulator of epithelial-to-mesenchyme transition, modulates snail’s subcellular localization and functions. Cancer research. 2005; 65(8):3179-3184.
75. Zheng H, Shen M, Zha YL, Li W, Wei Y, Blanco MA, Ren G, Zhou T, Storz P, Wang HY and Kang Y. PKD1 phosphorylation-dependent degradation of SNAIL by SCF-FBXO11 regulates epithelial-mesenchymal transition and metastasis. Cancer cell. 2014; 26(3):358-373.
76. Ko H, Kim HS, Kim NH, Lee SH, Kim KH, Hong SH and Yook JI. Nuclear localization signals of the E-cadherin transcriptional repressor Snail. Cells, tissues, organs. 2007; 185(1-3):66-72.
77. Vega S, Morales AV, Ocana OH, Valdes F, Fabregat I and Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes & development. 2004; 18(10):1131-1143.
78. Gupta GP and Massague J. Cancer metastasis: building a framework. Cell. 2006; 127(4):679-695.
79. Aktas B, Tewes M, Fehm T, Hauch S, Kimmig R and Kasimir-Bauer S. Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. BCR. 2009; 11(4):R46.
80. Armstrong AJ, Marengo MS, Oltean S, Kemeny G, Bitting RL, Turnbull JD, Herold CI, Marcom PK, George DJ and Garcia-Blanco MA. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. MCR. 2011; 9(8):997-1007.
81. Labelle M, Begum S and Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer cell. 2011; 20(5):576-590.
82. Wicki A, Mandala M, Massi D, Taverna D, Tang H, Hemmings BA and Xue G. Acquired Resistance to Clinical Cancer Therapy: A Twist in Physiological Signaling. Physiological reviews. 2016; 96(3):805-829.
83. Singh A and Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010; 29(34):4741-4751.
84. Dave B, Mittal V, Tan NM and Chang JC. Epithelial-mesenchymal transition, cancer stem cells and treatment resistance. BCR. 2012; 14(1):202.
85. Voulgari A and Pintzas A. Epithelial-mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochimica et biophysica acta. 2009; 1796(2):75-90.
86. Zhao X, Wang Y, Deng R, Zhang H, Dou J, Yuan H, Hou G, Du Y, Chen Q and Yu J. miR186 suppresses prostate cancer progression by targeting Twist1. Oncotarget. 2016. doi: 10.18632/oncotarget.8887.
87. Li LZ, Zhang CZ, Liu LL, Yi C, Lu SX, Zhou X, Zhang ZJ, Peng YH, Yang YZ and Yun JP. miR-720 inhibits tumor invasion and migration in breast cancer by targeting TWIST1. Carcinogenesis. 2014; 35(2):469-478.
88. Wang Y, Sun B, Zhao X, Zhao N, Sun R, Zhu D, Zhang Y, Li Y, Gu Q, Dong X, Wang M and An J. Twist1-related miR-26b-5p suppresses epithelial-mesenchymal transition, migration and invasion by targeting SMAD1 in hepatocellular carcinoma. Oncotarget. 2016; 7(17):24383-24401. doi: 10.18632/oncotarget.8328.
89. Sun T, Fu J, Shen T, Lin X, Liao L, Feng XH and Xu J. The Small C-terminal Domain Phosphatase 1 Inhibits Cancer Cell Migration and Invasion by Dephosphorylating Phospho-Ser68-Twist1 to Accelerate Twist1 Degradation. The Journal of biological chemistry. 2016.
90. Li CW, Xia W, Lim SO, Hsu JL, Huo L, Wu Y, Li LY, Lai CC, Chang SS, Hsu YH, Sun HL, Kim J, Yamaguchi H, et al. AKT1 Inhibits Epithelial-to-Mesenchymal Transition in Breast Cancer through Phosphorylation-Dependent Twist1 Degradation. Cancer research. 2016; 76(6):1451-1462.
91. Tran PT, Shroff EH, Burns TF, Thiyagarajan S, Das ST, Zabuawala T, Chen J, Cho YJ, Luong R, Tamayo P, Salih T, Aziz K, Adam SJ, et al. Twist1 suppresses senescence programs and thereby accelerates and maintains mutant Kras-induced lung tumorigenesis. PLoS genetics. 2012; 8(5):e1002650.
92. Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S, Maestro R, Voeltzel T, Selmi A, et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer cell. 2008; 14(1):79-89.
93. Xue G and Hemmings BA. PKB/Akt-dependent regulation of cell motility. Journal of the National Cancer Institute. 2013; 105(6):393-404.
94. Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, Natesan S and Brugge JS. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. The Journal of cell biology. 2005; 171(6):1023-1034.
95. Iliopoulos D, Polytarchou C, Hatziapostolou M, Kottakis F, Maroulakou IG, Struhl K and Tsichlis PN. MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Science signaling. 2009; 2(92):ra62.
96. Gajula RP, Chettiar ST, Williams RD, Nugent K, Kato Y, Wang H, Malek R, Taparra K, Cades J, Annadanam A, Yoon AR, Fertig E, Firulli BA, et al. Structure-function studies of the bHLH phosphorylation domain of TWIST1 in prostate cancer cells. Neoplasia. 2015; 17(1):16-31.
97. Sanidas I, Polytarchou C, Hatziapostolou M, Ezell SA, Kottakis F, Hu L, Guo A, Xie J, Comb MJ, Iliopoulos D and Tsichlis PN. Phosphoproteomics screen reveals akt isoform-specific signals linking RNA processing to lung cancer. Molecular cell. 2014; 53(4):577-590.
98. Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M and Hung MC. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nature cell biology. 2004; 6(10):931-940.
99. Aceto N, Sausgruber N, Brinkhaus H, Gaidatzis D, Martiny-Baron G, Mazzarol G, Confalonieri S, Quarto M, Hu G, Balwierz PJ, Pachkov M, Elledge SJ, van Nimwegen E, et al. Tyrosine phosphatase SHP2 promotes breast cancer progression and maintains tumor-initiating cells via activation of key transcription factors and a positive feedback signaling loop. Nature medicine. 2012; 18(4):529-537.
100. Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS and Kalluri R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015; 527(7579):525-530.
101. Mitra A, Mishra L and Li S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget. 2015; 6(13):10697-10711. doi: 10.18632/oncotarget.4037
102. Wilson C, Nicholes K, Bustos D, Lin E, Song Q, Stephan JP, Kirkpatrick DS and Settleman J. Overcoming EMT-associated resistance to anti-cancer drugs via Src/FAK pathway inhibition. Oncotarget. 2014; 5(17):7328-7341. doi: 10.18632/oncotarget.2397
103. Marcucci F, Stassi G and De Maria R. Epithelial-mesenchymal transition: a new target in anticancer drug discovery. Nature reviews Drug discovery. 2016; 15(5):311-325.