
Introduction
Multifactorial diseases are the final result of the interaction between genetic susceptibility and environmental factors in which a clear hereditary pattern is not found. This complexity causes difficulties in the risk evaluation, diagnosis and treatment of these diseases. Cancer, one of the most prevalent multifactorial diseases, is characterized by the lost of physiological control and the malignant transformation of cells that acquire functional and genetic abnormalities, leading to tumor development and progression. In some cases, cancer cells have the ability to invade other tissues resulting in metastasis, the major cause of death from cancer. According to the most recent data released by the World Health Organization (WHO) in 2012, more than 14 million of new cancer cases were diagnosed, and 8.2 million cancer deaths and 32.4 million people living with cancer (within 5 years of diagnosis) were registered worldwide [1]. The most common cancers by primary site location were lung, prostate and colorectal in men, and breast, colorectal and cervix uteri in women [1].
Tumor cells present common biological capabilities sequentially acquired during the development of cancer that are considered essential to drive malignancy and known as the hallmarks of cancer [2]. These hallmark capabilities include sustaining proliferative signaling, evading growth suppressors, avoiding immune destruction, enabling replicative immortality, activating invasion and metastasis, inducing angiogenesis, resisting cell death and reprogramming cellular metabolism. In addition, there are two consequential characteristics of tumorigenesis that enable the acquisition of the hallmarks of cancer. The most prominent is the development of genomic instability and mutability, which endow tumor cells with genetic alterations that can orchestrate tumor progression. The second one involves the tumor-promoting inflammation by innate immune cells, which in turn serve to support multiple hallmark capabilities [2].
Non-transformed cells tightly regulate the mitogenic signaling that command cell growth and division in order to maintain a balance between cell proliferation and death. Accordingly, the dysregulation of the signaling pathways that regulate the progression through cell cycle, cell survival and metabolism may lead to malignant transformation. It is worth noting that neoplastic transformation requires not only the alteration of proliferative stimuli but also the disruption of mechanisms that prevent unrestrained proliferation such as programmed cell death (apoptosis) or negative-feedback signaling [3]. Likewise, the cooperative activation of oncogenes (genes that promote cell growth, proliferation and survival) and/or inactivation of tumor suppressor genes (genes that restrain cell growth and proliferation, promote DNA repair or trigger apoptosis) are involved in tumor development [3, 4]. Oncogenes can be activated through several mechanisms including upregulated transcriptional expression, increased stability of mutant proteins, altered functionality of proteins and abnormal recruitment or subcellular localization of gene products through interaction with aberrantly expressed or mutant binding partners [3, 5]. The products of oncogenes comprise transcription factors (e.g. c-MYC, hereafter referred to as MYC), growth factor receptors (e.g. EGFR), signal transduction proteins (e.g. RAS and PI3K), serine-threonine protein kinases (e.g. Akt, mTOR, CDK4 and CDK6) and inhibitors of apoptosis (e.g. BCL2) [5]. On the other hand, tumor suppressor genes encode proteins that inhibit cell division and cell proliferation (e.g. RB, p53, p16INK4a, PTEN), stimulate cell death (e.g. caspase 8 and p53) and repair damaged DNA (e.g. MSH2, MSH6, ATM and ATR) [6].
Accumulation of genetic alterations is associated with tumor evolution, which includes single nucleotide mutations and also whole-chromosomal changes [7-9]. In addition, epigenetic mechanisms including histone modifications, DNA methylation and non-coding RNAs are involved in carcinogenesis [10, 11]. In fact, tumors often display aberrant methylation patterns such as hypermethylation on the promoters of tumor suppressor genes causing transcriptional repression, and hypomethylation of oncogenes supporting their activation (reviewed in [11, 12]). Epigenetic modifications have been reported to regulate the Warburg effect and coordinate the overall cellular metabolism, including the pentose phosphate pathway and other pathways for sugar, lipid and amino acid metabolism, by affecting several metabolic enzyme activities [13-16]. Remarkably, oxidative stress is involved with both genetic and epigenetic modifications, playing an important role in carcinogenesis [10, 17].
Metabolic reprogramming of tumor cells
Metabolism is the term that is used to describe the integrated network of chemical reactions involved in sustaining growth, proliferation and survival of cells and organisms. These reactions are catalyzed by tightly regulated enzymes, which sense environmental cues and provide energy, reducing power and macromolecules to supply the cellular needs. Metabolic reactions can be classified into catabolic pathways that produce energy (adenosine triphosphate, ATP) through the breakdown of molecules, and anabolic pathways that synthesize molecules through energy-consuming processes. The metabolic network is regulated by signaling pathways that respond to the specific cellular needs which, in turn, may vary depending on the cell type and proliferative state.
Despite the fact that there are several metabolic similarities between tumor and highly proliferating non-transformed cells (reviewed in [18]), oncogenic regulation and tumor microenvironment have a distinctive influence on the metabolic reprogramming of cancer cells. In particular, tumor cells switch their core metabolism to meet the increased requirements of cell growth and division. Indeed, tumor metabolic reprogramming involves the enhancement of key metabolic pathways such as glycolysis, pentose phosphate pathway, glutaminolysis and lipid, nucleic acid and amino acid metabolism [19] (Figure 1). Thus, activation of oncogenic signaling pathways adapts tumor cells metabolism to the dynamic tumor microenvironment, where nutrient and oxygen concentrations are spatially and temporally heterogeneous [20, 21]. The dependencies on specific metabolic substrates such as glucose or glutamine exhibited by tumor cells are determined by the alterations in their oncogenes and tumor suppressor genes. For instance, MYC-transformed cells display addiction to glutamine as a bioenergetic substrate and are sensitive to inhibitors of glutaminolysis [22]. Accordingly, the characterization of the metabolic reprogramming of cancer cells and its connection with oncogenic signaling is a promising approach to identify novel molecular-targeted strategies in cancer therapy.
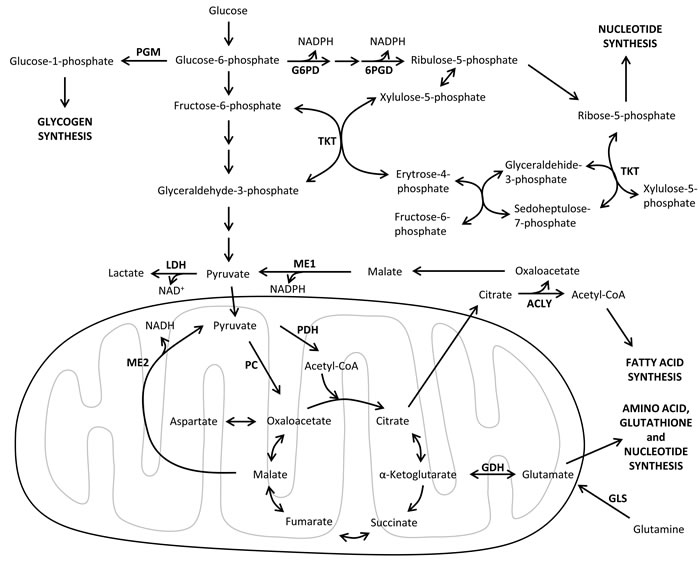
Figure 1: Major metabolic pathways involved in tumor metabolic reprogramming. An overview of the main catabolic and anabolic metabolic pathways supporting tumor cell growth and survival. Enzymes are shown in bold. 6PGD, 6-phosphogluconate dehydrogenase; ACLY, ATP citrate lyase; CoA, coenzyme A; GLS, glutaminase; GDH, glutamate dehydrogenase; G6PD, glucose-6-phosphate dehydrogenase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; LDH, lactate dehydrogenase; ME1, malic enzyme 1 cytoplasmic form; ME2, malic enzyme 2 mitochondrial form; NAD+, nicotinamide adenine dinucleotide oxidized form; NADH, nicotinamide adenine dinucleotide reduced form; NADPH, nicotinamide adenine dinucleotide phosphate reduced form; PC, pyruvate carboxylase; PDH, pyruvate dehydrogenase; PGM, phosphoglucomutase.
Glycolysis and the Warburg effect
Glycolysis is the metabolic pathway by which glucose and other sugars are metabolized to pyruvate in an oxygen-independent manner to generate energy in the form of ATP and intermediates, which are used as precursors for the biosynthesis of macromolecules [23]. Under physiologic oxygen concentrations, pyruvate enters the mitochondria to be oxidized through an oxygen-dependent process known as oxidative phosphorylation (OXPHOS), which couples the oxidation of metabolites and the electron transport chain (ETC) with ATP production, being also a potential source of reactive oxygen species (ROS) [20].
The first metabolic phenotype observed in tumor cells was described by Otto Warburg as a shift from oxidative phosphorylation to aerobic glycolysis to generate lactate and ATP even in presence of oxygen, which is known as the Warburg effect [24, 25]. Therefore, cancer cells convert most incoming glucose to lactate rather than entering in the mitochondria to be oxidized through oxidative phosphorylation [26]. Initially, it was believed that the Warburg effect resulted from defects in the mitochondrial function of cancer cells. However, this effect is also exhibited by tumor cells with intact and functional mitochondria, suggesting that their preference for glycolysis might confer benefits on them such as reduced levels of ROS, high production of metabolic intermediates for macromolecular biosynthesis and acidification of extracellular microenvironment due to lactate excretion [27, 28]. It is worth noting that the ATP produced per molecule of glucose catabolized through glycolysis is considerably less efficient than through oxidative phosphorylation (2 versus 31-38 molecules of ATP [29], respectively), causing tumor cells to greatly increase both the rate of glucose uptake and glycolysis to sustain their increased energetic, biosynthetic and redox needs [30]. Conveniently, the high glycolytic rates displayed by cancer cells allow their visualization by 18F-deoxyglucose positron emission tomography (FDG-PET) and assist tumor detection, prevention and treatment [31].
Over the past decade, numerous studies and reviews have supported the hypothesis that the Warburg effect can be explained by the alterations in multiple signaling pathways resulting from mutations in oncogenes and tumor suppressor genes [21, 28, 32-35]. The complex network of mechanisms leading to the Warburg phenomenon includes mitochondrial changes, upregulation of rate-limiting enzymes in glycolysis involving specific isoforms such as M2 pyruvate kinase and hexokinase 2, intracellular pH regulation, and hypoxia-induced switch to anaerobic metabolism (reviewed in [35]). The enhanced glycolytic rate can be sustained through the overexpression of glucose transporters [36] and several key glycolytic enzymes [37] mediated by specific activated oncogenes (e.g. PI3K and MYC) and transcription factors (e.g. HIF1), contributing to the acquisition of the Warburg effect and maintaining tumor cell growth and survival [21, 28, 33]. Likewise, loss-of-function mutations in tumor suppressor TP53 (encoding p53) also contribute to the Warburg effect, since they prevent i) p53-mediated transcriptional repression of glucose transporters GLUT1 and GLUT4; ii) activation of cytochrome c oxidase assembly protein (SCO2) expression, which promotes OXPHOS; and iii) upregulation of TP53-induced glycolysis and apoptosis regulator (TIGAR) expression, which reduces the intracellular concentration of the glycolytic activator fructose-2,6-bisphosphate [20, 38].
Interestingly, the metabolic switch in tumor cells has a key role in the establishment of many other cancer hallmarks [19]. In fact, some metabolic enzymes have been described as multifaceted proteins which can directly regulate transcription, glucose homeostasis and resistance to cell death [39, 40]. For example, hexokinase 2 isoform (HK2), which catalyzes the rate-limiting first step of glycolysis, plays a key role for the Warburg effect in cancer [41-43]. Specifically, HK2 bounds to mitochondria and is recognized as a signaling component controlling cellular growth, preventing mitochondrial apoptosis and enhancing autophagy [44, 45]. The competitive binding of HK2 to the voltage-dependent anion channel (VDAC) in the outer mitochondrial membrane prevents the union of VDAC with pro-apoptotic Bax, inhibiting cytochrome c release from mitochondria and avoiding apoptosis after Bax activation [44]. Therefore, targeting multifunctional metabolic enzymes may restore the susceptibility of tumor cells to cell death, offering new options for cancer therapy.
Pentose phosphate pathway
Pentose phosphate pathway (PPP) is one of the main metabolic pathways that enables tumor cell proliferation by regulating the flux of carbons between nucleic acid synthesis and lipogenesis to support DNA replication and RNA production. DNA and RNA nucleic acids are polymers composed by combinations of four different nucleotides which in turn are constituted by an organic base (purine, in the case of the nucleotides adenine and guanine, or pyrimidine, in the case of cytosine, thymine, and uracil), a pentose sugar (ribose for RNA or deoxyribose for DNA) and one or more phosphate groups. The pentose phosphate is mainly obtained through the PPP, which also generates nicotinamide adenine dinucleotide phosphate (NADPH). NADPH is an essential cofactor for providing reducing equivalents for lipid and amino acid biosynthesis, and for modulating oxidative stress through the maintenance of the reduced glutathione (GSH) pool [46]. The association between upregulation of PPP and tumor cell proliferation is been extensively studied, as PPP plays a pivotal role in allowing tumor cells to meet their anabolic demands and counteract oxidative stress [47-49].
PPP is divided into the oxidative branch and the non-oxidative branch. The oxidative branch catalyzes the irreversible transformation of glucose-6-phosphate into ribose-5-phosphate (R5P), yielding NADPH. The non-oxidative branch is a reversible pathway that interconverts R5P and glycolytic intermediaries. The enzymes that mainly regulate the PPP are glucose-6-phosphate dehydrogenase (G6PD) in the oxidative branch and transketolase (TKT) in the non-oxidative branch [50-52]. Several oncogenic signaling pathways promote G6PD activation by post-translational mechanisms [46], while tumor suppressor p53 directly inhibits G6PD and the PPP [48]. PPP is coordinated with cell cycle since proliferating cells increase G6PD activity during late G1 and S phases [53]. Moreover, the activation of the SCF ubiquitin ligase by its interaction with the protein-b-transduction repeat-containing protein (b-TrCP) allows the recognition of PFKFB3 and its proteasome degradation during S phase [54, 55], promoting the shuttling of glycolytic substrates through the PPP and increasing the production of NADPH and R5P to allow S phase progression.
Lipid metabolism
Triacylglycerides, phosphoglycerides, sterols and sphingolipids are hydrophobic or amphipathic molecules known as lipids. Fatty acids are long hydrocarbon chains with a carboxy-terminal group that constitute the main component of triacylglycerides and phosphoglycerides, being also present in sphingolipids and sterol esters. While triacylglycerides are used as energy storage units, phosphoglycerides, sterols and sphingolipids are major structural components of plasma membranes. Lipids are also involved in signal transduction and participate in the regulation of cell growth, proliferation, differentiation, survival, apoptosis, membrane homeostasis, motility and drug resistance [56, 57].
Tumor metabolic reprogramming involves an increase in lipid biosynthesis to supply the building blocks for membrane formation and sustain the high proliferative rate of tumor cells. Distinctively, tumor cells mainly activate and thrive on de novo lipid biosynthesis, while most non-transformed cells rely on extracellular lipids. Oncogenic signaling enhances lipogenesis through the increase of precursors for fatty acids synthesis (i.e. promoting glucose and glutamine transport, glycolysis, PPP and anaplerosis) and the upregulation of many lipogenic enzymes such as ATP citrate lyase (ACLY), fatty acid synthase (FASN) and acetyl-CoA carboxylase (ACC) [58-61]. The acetyl groups for fatty acids biosynthesis are provided by mitochondrial citrate, which is exported to the cytosol where ACLY catalyzes its conversion into acetyl-CoA and oxaloacetate [62]. Then, malate dehydrogenase (MDH) and malic enzyme (ME) can produce pyruvate from oxaloacetate, yielding part of the NADPH required for fatty acid biosynthesis. In addition, lipid biosynthesis is also connected to other pathways that generate NADPH, such as the oxidative branch of the PPP. Next, acetyl-CoA is converted to malonyl-CoA by ACC, and both acetyl and malonyl groups are condensed through a cyclical series of reactions by FASN, resulting in long-chain saturated fatty acids, predominantly palmitate. Further elongation and desaturation of de novo synthesized saturated fatty acids can be obtained through the action of elongases and desaturases [56, 63]. On the other hand, the mitochondrial degradation of fatty acids through β-oxidation releases large amounts of ATP and generates ROS through the TCA cycle and the oxidative phosphorylation [56, 57].
Sterol regulatory element-binding proteins (SREBPs) transcription factors regulate the expression of most enzymes involved in the synthesis of fatty acids and cholesterol. In turn, SREBPs are negatively regulated by tumor suppressors such as p53, pRB and AMPK, and activated by oncogenes such as PI3K and Akt. For instance, besides promoting glycolysis, Akt upregulates the expression of the lipogenic enzymes through activation and nuclear translocation of SREBP [64], and positively regulates ACLY by direct phosphorylation [65], linking enhanced glycolysis with increased lipogenesis [63, 66]. Therefore, targeting lipogenic pathways is thought to be a promising strategy for cancer therapy, as lipogenic enzymes are found to be upregulated or activated in tumor cells to satisfy their increased demand for lipids [57, 58].
Amino acid metabolism
Amino acids are organic compounds containing a specific side chain and both amino and carboxyl groups that enable them to undergo polymerization to form proteins. In addition, amino acids can be metabolized as a source of carbon and nitrogen for biosynthesis. There are 20 different amino acids, 11 of which can be endogenously synthesized by mammal cells while the remainder are known as essential amino acids, which must be obtained from external sources. In fact, amino acids have a pivotal role in supporting proliferative metabolism and are required for cell survival. It is not surprising that cells have developed an amino acid sensing system through the mechanistic target of rapamycin (mTOR) signaling to determine whether there are sufficient amino acids available for protein biosynthesis. Specifically, leucine, glutamine and arginine serve as critical signaling molecules that activate mTOR pathway [67, 68]. In response to amino acid deficiency, inhibition of mTOR rapidly suppress protein synthesis and induce autophagy, in order to maintain a free amino acid pool which may be required during prolonged amino acid limitation [69].
Non-essential aminoacids can be synthesized from glycolytic intermediates such as 3-phosphoglycerate, which is the precursor for serine, or pyruvate, that can be converted to alanine. In addition, TCA intermediates like oxaloacetate and α-ketoglutarate can generate aspartate, asparagine and glutamate. Moreover, glutamate can be converted to L-glutamate-5-semialdehyde (GSA) and 1-pyrroline-5-carboxylate (P5C), which are further converted to ornithine and proline, respectively [70]. Then, ornithine can enter the urea cycle and produce arginine. Also, serine can generate glycine and contribute to the synthesis of cysteine [71].
Highly proliferating cells, like tumor cells, consume essential and non-essential amino acids from external sources since the capacity of endogenous synthesis is not sufficient to fulfill their amino acidic increased needs [72]. However, most amino acids are hydrophilic molecules that require selective transport proteins to cross the cell membrane. Accordingly, four amino acid transporters (SLC1A5 [22, 73], SLC7A5 [73], SLC7A11 [74] and SLC6A14 [75]) have been found to be overexpressed in cancer cells in a MYC-dependent manner or through miR-23a repression mediated by MYC to increase the uptake of amino acids and meet their growing demands [72]. Interestingly, the functional coupling of SLC1A5 and SLC7A5 glutamine transporters suggests that enhanced glutamine metabolism in tumor cells can contribute to drive tumor growth through activation of mTOR [68].
In tumor cells, the consumption of some amino acids (specially non-essential amino acids) greatly exceeds the requirements for protein biosynthesis, suggesting their use as intermediates in metabolism by providing one carbon units, replenishing the TCA cycle or synthesizing fatty acids, nucleotides and other amino acids [71]. For example, glutamine, glycine and aspartate are required for nucleotide biosynthesis, while serine and glycine play an essential role in a one-carbon metabolism, generating precursors for the biosynthesis of lipids, nucleotides and proteins, regulating the redox status and participating in protein and nucleic acid methylation [76, 77]. The conversion of serine to glycine can be catalyzed either by the cytosolic or mitochondrial serine hydroxymethyltransferase (SHMT1 and SHMT2, respectively). Interestingly, the metabolic activity of SHMT2 has been shown to strongly correlate with the rates of proliferation across the NCI60 cancer cell collection [78]. In fact, SHMT2 has been suggested as fundamental to sustain cancer metabolism by fuelling heme biosynthesis and thus oxidative phosphorylation [79].
It is worth noting that the reactions catalyzing the degradation of proline produce significant amounts of ROS. The first step of proline degradation is catalyzed by the mitochondrial proline dehydrogenase (PRODH), which is a tumor suppressor that inhibits proliferation and induces apoptosis [70, 80]. This mitochondrial enzyme is linked to the electron transport chain through complex III, being shown as a source of ROS generation. In addition, P5C and proline can act as a redox couple, carrying reducing potential into and oxidizing potential out of the mitochondria by the combined activities of mitochondrial PRODH and the cytosolic form of P5C reductase (PYCR), which preferably uses NADPH [70, 80, 81].
It is worth noting that glutamine is the amino acid presenting the most prominent role in tumor metabolism. Accordingly, some tumor cells have been reported to exhibit dependence on glutamine for survival [22, 82].
Mitochondrial metabolism
Mitochondrial function is essential for cancer cells as it is involved in numerous crucial cellular processes such as ATP generation, regulation of programmed cell death, and regulation of signal transduction pathways through ROS production, modulation of cytosolic calcium levels and trafficking of small metabolites. Indeed, impairment of mitochondrial function and reduction of mitochondrial biogenesis greatly suppresses tumor formation, growth and proliferation [63, 83, 84]. Conversely, enhancement of mitochondrial biogenesis is advantageous for tumor cells [63, 85]. On the other hand, alterations in mitochondrial function can lead to several diseases including cardiovascular dysfunctions, muscular degeneration and cancer [83, 86].
In the presence of oxygen, oxidative phosphorylation (OXPHOS) is the most efficient mechanism for synthesizing ATP [29]. OXPHOS is coupled to the oxidation of reduced nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) through the electron transport chain. The mitochondrial respiratory chain, located in the mitochondrial inner membrane, comprises four complexes (I to IV) that are responsible for the oxidation of the reducing equivalents in the form of NADH or FADH2 and the reduction of molecular oxygen (final electron acceptor) to water. This process is coupled to the pumping of protons into the mitochondrial intermembrane space, resulting in a proton gradient that is used by the ATPase (complex V) to produce ATP [87] (Figure 2).
Among the metabolic pathways that take place in the mitochondria, the tricarboxylic acid (TCA) cycle is a route of pivotal importance for the entire cellular metabolism and, in particular, for oxidative metabolism. Remarkably, TCA cycle provides precursors for the biosynthesis of lipids, nucleic acids and proteins, as well as reducing equivalents (NADH and FADH2) for ATP production (Figure 2). Mutations in several genes that encode enzymes of the TCA cycle including isocitrate dehydrogenase [88, 89], succinate dehydrogenase and fumarate hydratase [90] are associated with some tumor types, leading to the dysfunction of the TCA cycle and the accumulation of its substrates [86, 91, 92]. Interestingly, it has been shown that increased levels of TCA cycle intermediates fumarate and succinate can affect α-ketoglutarate-dependent histone and DNA demethylases, HIF stabilization, and cellular responses to O2 depletion [90, 93]. Isocitrate dehydrogenases (IDHs) catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate, which is required as a substrate for numerous dioxygenases, including histone demethylases, prolyl hydroxylases, collagen prolyl-4-hydroxylases, and the TET family of 5-methylcytosine hydroxylases [94, 95]. There are three IDH enzymes; IDH1 is found in the cytosol and peroxisome, while IDH2 and IDH3 isoforms are localized in the mitochondria. Mutations targeting IDH1 and IDH2 result in loss of their native enzymatic activities and lead to the production of 2-hydroxyglutarate, a metabolite that can competitively inhibit α-ketoglutarate-dependent dioxygenases and is associated with tumorigenesis [95-97].
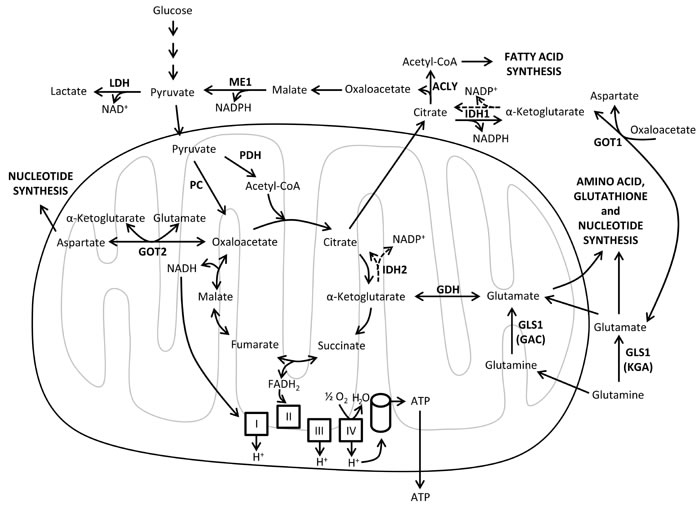
Figure 2: Mitochondrial metabolism. Schematic representation of the biosynthetic and bioenergetic reactions of the TCA cycle and the oxidative phosphorylation (OXPHOS). OXPHOS is coupled to the oxidation of NADH and FADH2, which are produced in the TCA cycle, via the electron transport chain (ETC, also known as the mitochondrial respiratory chain). The ETC comprises four complexes (I to IV) that transfer electrons generating a gradient of protons (H+) in the mitochondrial intermembrane space, which is used by the ATPase (complex V) to produce ATP. Reductive carboxylation of α-ketoglutarate by IDH1 and IDH2 produces citrate (dashed arrows). ACLY, ATP citrate lyase; ATP, adenosine triphosphate; CoA, coenzyme A; FADH2, flavin adenine dinucleotide reduced form; GAC, glutaminase C; GDH, glutamate dehydrogenase; GLS1, glutaminase 1; GOT1, glutamic-oxaloacetic transaminase 1 cytoplasmic form; GOT2, glutamic-oxaloacetic transaminase 2 mitochondrial form; IDH1, isocitrate dehydrogenase cytoplasmic form; IDH2, isocitrate dehydrogenase mitochondrial form; KGA, kidney (K-type) glutaminase; LDH, lactate dehydrogenase; ME1, malic enzyme 1 cytoplasmic form ; NAD+, nicotinamide adenine dinucleotide oxidized form; NADH, nicotinamide adenine dinucleotide reduced form; NADP+, nicotinamide adenine dinucleotide phosphate oxidized form; NADPH, nicotinamide adenine dinucleotide phosphate reduced form; PC, pyruvate carboxylase; PDH, pyruvate dehydrogenase.
Glutamine metabolism
Glutamine is the most abundant amino acid in plasma and in intracellular pools, being consumed at significantly higher rates than other amino acids by tumor cells [82]. Glutamine plays several cellular key roles as a nitrogen donor for nucleotide and protein synthesis, as a carbon source for energy production and lipid biosynthesis, and as a precursor for some non-essential amino acids and the antioxidant GSH biosynthesis [82, 98]. Despite being a non-essential amino acid, glutamine is crucial for the proliferation of most cells and for the viability of some tumor cells that have developed glutamine dependence [99].
The expression levels of oncogenes (e.g. MYC [22, 100], mTOR [101] and KRAS [102]) and tumor suppressors (e.g. SIRT4 [103] and TP53 [104]) are decisive to regulate glutamine metabolism [92, 105], to the extent that tumor genetics can dictate cellular dependence on glutamine for survival [98]. For instance, tumor cells overexpressing MYC reprogram their mitochondrial metabolism to depend on glutamine for the maintenance of cell viability, mitochondrial integrity and TCA cycle anaplerosis, triggering cellular addiction to glutamine and displaying increased sensitivity to glutamine deprivation [22, 100].
In addition to glycolysis, many tumor cells also rely on glutamine to fulfill their bioenergetic and metabolic needs. Indeed, glutamine catabolism is the source of many precursors for major anaplerotic processes such as the TCA cycle. Cells requiring de novo lipid biosynthesis, like tumor cells, divert citrate from the TCA cycle to produce lipogenic acetyl-CoA. The depletion of citrate from the TCA cycle creates a need for anaplerotic replenishment of the cycle, which can be provided through oxidative metabolism of glutamine [106]. Oxidation of glutamine in the mitochondria begins with its conversion to glutamate catalyzed by glutaminase (GLS). Glutaminase is a pivotal enzyme in the regulation of glutamine metabolism in tumor cells which has recently gathered some attention as a promising target for cancer therapy [107-109]. There are three mammalian glutaminase isoforms; kidney (K-type) glutaminase (KGA) and glutaminase C (GAC) are encoded by GLS and referred to as GLS1, while liver (L-type) glutaminase (LGA) is encoded by GLS2 and usually known as GLS2 [110]. Glutamate can be converted to α-ketoglutarate by either glutamate dehydrogenase (GDH) or transaminases, to feed the TCA cycle (Figure 2). In addition, glutamate can serve as a precursor of GSH and non-essential amino acids such as aspartate, alanine, proline and arginine. Interestingly, α-ketoglutarate levels are determinant for the regulation of HIF1α degradation through prolyl hydroxylase (PHD) sensing pathway [67, 94]. Furthermore, glutaminolysis and α-ketoglutarate are also involved in the activation of mTOR signaling [68, 111]. Glutamine carbons can exit the TCA cycle in the form of malate, which can be converted to pyruvate by malic enzyme (ME) with NADPH generation [112]. Both glutamine-derived NADPH and GSH production allow tumor cells to reduce the oxidative stress associated with mitochondrial respiration and rapid cell proliferation.
It is worth noting that glutamine utilization as a respiratory substrate through the TCA cycle produces NADH and FADH2 that provide electrons for the mitochondrial electron transport chain to generate ATP (Figure 2). Remarkably, glycolytic contribution to total ATP synthesis in tumor cells differs widely depending on cell type, from over 60% to less than 1%, with a mean contribution of 17±18% in the tested cell lines [113]. These results are confirmed by a flux balance analysis across the NCI-60 cell lines [78, 114] which shows that oxidative phosphorylation contributes to 70-84% of the total cellular ATP production [115]. Therefore, oxidative metabolism of glutamine is the major energetic source in many cancer cell lines.
Together, glucose and glutamine are the two principal nutrients to coordinately fuel the proliferation of tumor cells by supplying not only ATP but also key precursors for protein, lipid and nucleic acid biosynthesis (Figures 1 and 2). In fact, some cancer cells can switch their carbon source in response to nutrient availability. For example, glucose withdrawal increases GDH activity in MYC-transformed glioblastoma cells [116], while impairing the oxidative metabolism of glutamine by silencing glutaminase induces a compensatory anaplerotic mechanism catalyzed by pyruvate carboxylase (PC) that enables the use of glucose-derived pyruvate for anaplerosis [117]. However, the metabolic compensation adopted by tumor cells renders them absolutely dependent on the new upregulated pathways, opening new opportunities for cancer combined therapies. Therefore, the metabolic flexibility and compensatory abilities exhibited by some tumor cells have to be carefully considered when designing cancer therapies.
Glutamine reductive carboxylation
There are two different glutamine-dependent pathways for fatty acid biosynthesis. On the one hand, cells can oxidatively metabolize glutamine-derived α-ketoglutarate to citrate in the TCA cycle and subsequently transport it to the cytosol to generate oxaloacetate and lipogenic acetyl-CoA [62]. Likewise, malate produced from glutamine in the TCA cycle can generate pyruvate through the action of malic enzyme, which can be further metabolized to lipogenic acetyl-CoA. On the other hand, α-ketoglutarate obtained from glutamine can be directly converted to citrate by reductive carboxylation, especially in tumor cells under hypoxic conditions or when mitochondrial respiration is impaired, in order to sustain cell growth under these circumstances [118-121]. This reaction takes advantage of the reversible transformation catalyzed by aconitase and isocitrate dehydrogenase. The cytosolic NADP+/NADPH-dependent isocitrate dehydrogenase 1 (IDH1) is the main enzyme catalyzing the reversible reductive carboxylation of α-ketoglutarate to isocitrate and NADP+ [118] (Figure 2). Indeed, reductive carboxylation of glutamine provides a glucose-independent pathway to generate acetyl-CoA for biosynthesis, allowing cells to conserve glucose for the production of biosynthetic precursors that are specifically generated from glucose [118].
Oncogenic regulation of tumor metabolic reprogramming
Tumor metabolic reprogramming is a direct result of the re-engineering of intracellular signaling pathways that are altered by mutations in oncogenes and tumor suppressor genes. In fact, most cancers harbor activating mutations of oncogenes and/or inactivating mutations of tumor suppressor genes which determine the tumor metabolic phenotype and support tumorigenesis by giving to transformed cells a proliferative advantage over non-malignant cells. Several oncogenes including MYC, hypoxia inducible factor 1 (HIF1), phosphoinositide-3-kinase (PI3K), protein kinase B (PBK or Akt) and the mechanistic target of rapamycin (mTOR), have been known to be involved in the regulation of tumor metabolic reprogramming [5, 20, 92].
MYC as a master regulator of tumorigenesis
The MYC oncogene belongs to the MYC family of genes together with MYCN and MYCL. However, MYC is the only isoform ubiquitously expressed in a broad range of tissues, while MYCN and MYCL are normally only expressed during development [122]. MYC is a multi-functional transcription factor that exerts control over cell proliferation, cell cycle progression, cell growth, metabolism, apoptosis, differentiation and stress response through transcriptional regulation of its target genes [122, 123]. In fact, MYC binds to the promoter of 10-15% of all known genes, regulating both genes encoding proteins and those encoding non-coding RNA products of several functional classes [122, 124]. MYC expression is dysregulated in many human cancers by either chromosomal translocation or gene amplification. In addition, the expression and stability of MYC protein and MYC mRNA can also be dysregulated, promoting tumorigenesis through unrestricted cell proliferation, inhibition of cell differentiation, metabolic adaptation, angiogenesis, reduction of cell adhesion and genomic instability [122, 123, 125, 126].
To function as a transcription factor, MYC protein heterodimerizes with its binding partner MAX, forming an activated complex that recognizes E box sequences (CACGTG) and induces the transcription of its targets genes. MYC can also act as transcriptional repressor by binding to MIZ1 or SP1 transcription factors and interfering with their transcriptional activity [127]. It is worth noting that multiple genes that are repressed by MYC encode negative regulators of cell proliferation such as CDKN2B (encoding p15INK4b), CDKN2C (p18INK4c), CDKN1A (p21Cip1), CDKN1B (p27Kip1), and CDKN1C (p57Kip2) [127]. MAX can also bind to MAD1, MXI1, MAD3, MAD4 and MNT or form homodimers, repressing the transcriptional activation of MYC target genes [128].
MYC and metabolism
MYC is known to enhance glycolysis through the activation of glycolytic genes (such as HK2, GAPDH, ENO1 and PK, among others) and glucose transporters (SLC2A1, SLC2A2 and SLC2A4) [129, 130]. In addition, MYC promotes lactate production and export, increasing the gene expression of LDHA and lactate transporter MCT1 [126, 131, 132]. Figure 3 illustrates the main metabolic pathways regulated by MYC.
On the other hand, transformed cells exhibit increased MYC-dependent glutaminolysis and glutamine dependency [22, 73]. Indeed, MYC has been described as the main oncoprotein responsible for inducing a transcriptional program that promotes glutaminolysis and triggers cellular addiction to glutamine as a bioenergetic substrate [22]. This glutamine addiction leads tumor cells to reprogram intermediate metabolism for the maintenance of mitochondrial tricarboxylic acid (TCA) cycle integrity [22]. Moreover, high levels of MYC promote mitochondrial biogenesis and function, both increasing the rate of oxygen consumption and the energy production required for rapid cell proliferation [125, 133-135]. High glutaminolysis rate results in the robust production of NADPH, which is needed to fulfill the requirements for cell proliferation [22, 112]. In conditions of low glucose and oxygen availability, MYC-induced glutamine catabolism is important for cell survival [108]. Furthermore, cells with supraphysiological levels of MYC are more sensitive to inhibition of mitochondrial oxidative metabolism [136]. Moreover, MYC is also found to contribute to increase glutamine uptake by upregulation of the expression of glutamine transporters (ASCT2 [SLC1A5] and SLC7A5) [73, 85]. Importantly, MYC enhances glutaminolysis by transcriptionally repressing miR-23a and miR-23b (microRNA-23a/b), resulting in greater expression of their target protein, glutaminase (GLS1) [73]. In fact, GLS1 is the first enzyme in the glutaminolysis and catalyzes the conversion of glutamine to glutamate for its oxidation in the TCA cycle and also for protein or glutathione synthesis. It is worth mentioning that MYC can stimulate the use of the TCA cycle to generate intermediates for macromolecular synthesis using both glucose and glutamine as carbon source [98, 108]. However, it has been reported that cells presenting high MYC levels greatly rely on mitochondrial oxidative phosphorylation and increase glutaminolysis by 2- to 4-fold, while only moderately increasing glycolysis by 1.2-fold [137].
Additionally, MYC has been shown to activate nucleotide biosynthesis by inducing several gens involved in nucleotide metabolism including carbamoyl-phosphate synthase / aspartate carbamoyltransferase / dihydroorotase (CAD), CTP synthase 1 (CTPS) and ornithine decarboxylase (ODC) [129, 132, 138, 139]. Polyamine biosynthesis is also regulated by MYC since ornithine decarboxylase (ODC) (the rate-limiting enzyme in polyamine production) [138], adenosylmethionine decarboxylase (AMD1) and spermidine synthase (SRM) have E boxes in their regulatory region and are enhanced in MYC-expressing cells [132]. Furthermore, polyamines stimulate MYC transcription in a positive feedback loop [140, 141].
Moreover, MYC can redirect glycolytic flux from 3-phosphoglycerate for the synthesis of serine and glycine involving folate metabolism, which are essential for purine and thymidylate biosynthesis [85, 132, 142, 143]. MYC is also implicated in proline metabolism regulation by transcriptionally repressing proline oxidase/proline dehydrogenase (POX/PRODH) expression through upregulation of miR-23b*, and increasing the expression of the enzymes of proline biosynthesis pathway (P5C synthase, P5CS and P5C reductase 1, PYCR1) [70].
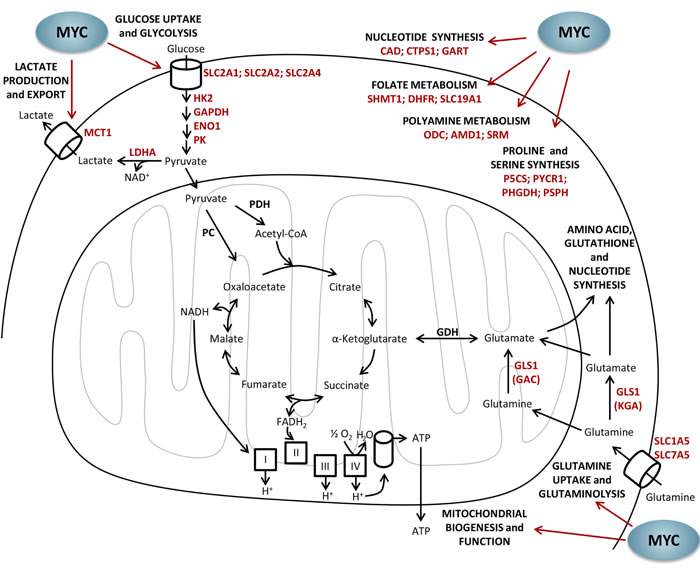
Figure 3: Metabolic regulation by MYC. MYC has a pivotal role in the metabolic reprogramming of tumor cells by enhancing glucose uptake and glycolysis, lactate production and export, glutamine uptake and glutaminolysis, mitochondrial biogenesis and oxidative phosphorylation, and nucleotide, folate, polyamine, proline and serine synthesis. AMD1, adenosylmethionine decarboxylase; ATP, adenosine triphosphate; CAD, carbamoyl-phosphate synthase/aspartate carbamoyltransferase/dihydroorotase; CoA, coenzyme A; CTPS1, cytidine triphosphate synthase 1; DHFR, dihydrofolate reductase; FADH2, flavin adenine dinucleotide reduced form; GAC, glutaminase C; GART, phosphoribosylglycinamide formyltransferase, phosphoribosylglycinamide synthetase, phosphoribosylaminoimida-zole synthetase; GDH, glutamate dehydrogenase; GLS1, glutaminase 1; KGA, kidney (K-type) glutaminase; LDHA, lactate dehydrogenase A; MCT1, monocarboxylate transporter/SLC16A1 solute carrier family 16 member 1; NAD+, nicotinamide adenine dinucleotide oxidized form; NADH, nicotinamide adenine dinucleotide reduced form; NADP+, nicotinamide adenine dinucleotide phosphate oxidized form; NADPH, nicotinamide adenine dinucleotide phosphate reduced form; ODC, ornithine decarboxylase; P5CS, Δ1-pyrroline-5-carboxylate synthetase; PHGDH, phosphoglycerate dehydrogenase; PC, pyruvate carboxylase; PDH, pyruvate dehydrogenase; PSPH, phosphoserine phosphatase; PYCR1, Δ1-pyrroline-5-carboxylate reductase 1; SHMT1, serine hydroxymethyltransferase 1; SLC1A5, solute carrier family 1 (neutral amino acid transporter) member 5; SLC2A1, solute carrier family 2 (facilitated glucose transporter) member 1; SLC2A2, solute carrier family 2 (facilitated glucose transporter) member 2; SLC2A4, solute carrier family 2 (facilitated glucose transporter) member 4; SLC7A5, solute carrier family 7 (amino acid transporter light chain, L system) member 5; SLC19A1, solute carrier family 19 (folate transporter) member 1; SRM, spermidine synthase.
MYC and cell cycle
The network of MYC target genes suggests its implication in the fulfillment of the metabolic requirements for cell cycle entry [136, 144]. In fact, one of the earliest observations after MYC discovery was its ability to promote cell proliferation and inhibit cell differentiation [129]. Remarkably, MYC overexpression in quiescent cells is sufficient to trigger cell cycle entry, reduce the requirement for growth factors, block cell cycle exit, and increase cell size [145, 146]. MYC promotes cell cycle progression by regulation of pivotal cell cycle control genes through transcriptional induction of CDKs and cyclins, and repression of CIP/KIP proteins. MYC mediates the increase of active cyclin-CDK complexes levels not only by upregulation of CDK1, CDK2, CDK4 (one of the principal MYC target genes [147]), CDK6, CCND1 (encoding cyclin D1), CCND2 (cyclin D2), CCND3 (cyclin D3), CCNE1 (cyclin E1), CCNE2 (cyclin E2), CCNA2 (cyclin A2) and CCNB1 (cyclin B1), but also by induction of CDC25A (CDKs phosphatase) and CDK activating kinase complex (CAK, through enhanced mRNA translation of its subunits, CDK7, cyclin H and MAT1), and repression of the CDK inhibitory kinase WEE1 through miR-221 activation [122, 126, 129, 145, 148]. Moreover, MYC abrogates the transcription of cell cycle checkpoint genes GADD45 and GADD153 [122, 148], and impairs the activity of the CDK inhibitors p27Kip1, p21Cip1 and p15INK4b through several mechanisms [122, 126, 129, 145]. One of the most studied mechanisms for p21Cip1 and p15INK4b MYC-mediated repression is the binding to MIZ1 and the blocking of its transcriptional activity [129, 145]. In contrast, MYC antagonizes p27Kip1 function by several parallel mechanisms such as induction of miR-221 and miR-222, activation of E2F transcription factors, increase of CDK4/6-cyclin D and CDK2-cyclin E complexes levels, and enhancement of the expression of several components of the SCF ubiquitin ligase complex [122, 145]. Last but not least, MYC further stimulates cell cycle progression by inducing genes directly involved in DNA replication including MCM (minichromosome maintenance), ORC (origin recognition complex), CDC6 (cell division cycle 6), TERT (telomerase reverse transcriptase) and the genes encoding three subunits of the APC/C (ANAPC5, CDC16 and CDC23) [145].
MYC regulation
Given its pivotal role on cell fate, MYC expression is tightly regulated at transcriptional, post-transcriptional and post-translational levels in non-transformed cells. Accordingly, dysregulation of MYC expression is one of the most common abnormalities in human diseases, being MYC overexpression frequently found in most human cancers. Remarkably, MYC oncogenic activation results from insertional mutagenesis, chromosomal translocation and gene amplification mechanisms, while most oncogenes are activated by mutations in their coding sequence [122].
MYC protein presents extremely short half-life (in the order of 20-30 minutes [149]) in the absence of mitogenic signals, but is transiently stabilized upon cell cycle entry and RAS activation, allowing its accumulation [150, 151]. RAS promotes MYC stability through RAF/MEK/ERK kinase cascade and via glycogen synthase kinase-3β (GSK-3β) inhibition by the PI3K/Akt pathway [150, 152]. MYC turnover is regulated by the ubiquitin proteasome pathway [149, 153] and is dependent on the phosphorylation of two highly conserved residues located near the N-terminal region of MYC, Thr58 and Ser62. These phosphorylation sites exert opposing control effects on MYC degradation [150]. ERK (extracellular receptor kinase) phosphorylates MYC on Ser62, promoting its protein accumulation, while phosphorylation of Thr58, which is mediated by GSK-3β but dependent on prior Ser62 phosphorylation, triggers MYC proteasomal degradation [150-152, 154]. Therefore, MYC phosphorylation at Ser62 has two opposite roles; MYC stabilization and accumulation, and activation of the subsequent phosphorylation at Thr 58, triggering MYC degradation. Interestingly, proteasome inhibition studies reveal that the accumulated poly-ubiquitinated MYC only exhibits phosphorylation on Thr58 [152, 154]. Since phosphorylation on Ser62 is required prior to Thr58 phosphorylation, the Ser62 phosphate is removed before MYC ubiquitination by protein phosphatase 2A (PP2A) action, contributing to MYC degradation [150, 152, 154].
In non-transformed cells, growth stimuli lead to RAS activation and MYC protein synthesis. However, when mitogenic signaling ends, RAS and PI3K activities decline and release GSK-3β from its negative regulation, activating its kinase activity and thus promoting MYC degradation by phosphorylation on Thr58 [154]. The ordered phosphorylation of Ser62 and Thr58 followed by Ser62 dephosphorylation allows a tight control of MYC protein levels. Hence, the disruption of the physiological regulation of MYC expression can lead to malignancy.
HIF1
The hypoxia inducible factors HIF1, HIF2 and HIF3 are the principal regulators of the transcriptional homeostatic responses to situations of limited availability of oxygen. HIF1 is ubiquitously expressed while HIF2 and HIF3 are only expressed in certain tissues [155]. Only HIF1 and HIF2 are further discussed in this section since HIF3 function is less well understood. The HIF factors are composed of an oxygen-dependent HIFα subunit and a constitutively expressed HIFβ subunit. HIF activity is tightly regulated by cycles of synthesis and oxygen-dependent proteasomal degradation. Indeed, HIFα subunits are continuously synthesized and their stability is regulated by oxygen availability [155]. Under normoxic conditions, HIFα subunits are hydroxylated on proline residues in the oxygen-dependent degradation (ODD) domain by prolyl hydroxylase enzymes (PHDs) and subsequently ubiquitinated by the tumor suppressor protein von Hippel-Lindau (VHL) prior to their degradation in the proteasome [155, 156] (Figure 4). Under hypoxic conditions, the reduced molecular oxygen levels decrease the activity of PHDs, which are further inactivated through the oxidation of the ferrous ion within their active sites by ROS released from inefficient mitochondrial respiration [157], thus preventing their interaction with VHL [156]. Consequently, stable HIFα subunits form heterodimers with HIFβ subunits and translocate to the nucleus, where they bind to specific consensus sequences (hypoxia response element, HRE) in the promoter of hypoxia-responsive genes for the transcriptional activation of the cellular adaptation to hypoxia [158].
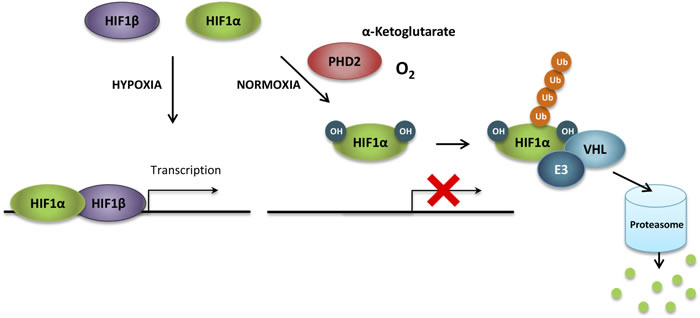
Figure 4: HIF1α regulation under normoxia. In the presence of oxygen and α-ketoglutarate, HIF1α subunits are hydroxylated on proline residues in the oxygen-dependent degradation (ODD) domain by prolyl hydroxylases (principally prolyl hydroxylase 2, PHD2). Prolyl hydroxylation is required for the binding of the von Hippel-Lindau protein (VHL), which recruits an ubiquitin ligase complex (E3) that ubiquitinates HIF1α. Ubiquitination marks HIF1α for proteasomal-mediated degradation. HIF, hypoxia inducible factor; OH, hydroxylation; Ub, ubiquitin.
Hypoxia and cancer
Solid tumors frequently develop hypoxia when highly proliferating tumor cells outgrow their vascular network, resulting in tumors with limited oxygen diffusion. In order to adapt to the hypoxic microenvironment and support cell survival, cells principally initiate response mechanisms through HIFα stabilization and accumulation, favoring angiogenesis, invasion and metabolic reprogramming [159, 160]. Accordingly, HIFα levels are increased in many human cancers and correlate with poor clinical prognosis [160]. It is worth mentioning that tumor cells can exhibit augmented levels of HIF1α under normoxic conditions, a phenomenon known as pseudo-hypoxia [159]. For example, induction of RAS or SRC oncogenic signaling promotes normoxic HIF1α accumulation through prolyl hydroxylation inhibition [161].
Regulation of HIF by prolyl hydroxylases
In humans, there are three different members of the prolyl hydroxylase family; PHD1, PHD2 and PHD3. However, only PHD2 has been confirmed to be involved in the oxygen regulation of HIF1α, while PHD1 and PHD3 display only partial additive effects on HIF1α stability [162]. These enzymes are good oxygen sensors since their affinity for oxygen is low with Km values from 230 to 250 µM, slightly above the concentration of oxygen in the air (200 µM) [163]. PHDs require α-ketoglutarate, oxygen and a prolyl residue as substrates, and iron and ascorbate as cofactors, to produce a hydroxyl-prolyl residue, succinate and CO2 [94]. Prolyl hydroxylation is required for the recognition and binding of VHL to the ODD domain, which recruits an ubiquitin ligase complex [158] (Figure 4). Chemical inhibitors of the activity of PHD, such as iron chelators (e.g. desferrioxamine, DFO) or competitors of α-ketoglutarate for binding at the hydroxylase (e.g. dimethyloxalylglycine, DMOG), prevent the hydroxylation of HIFα subunits, causing their accumulation and promoting the expression of HIF target genes [164]. Remarkably, the use of α-ketoglutarate as an electron donor in the reaction of hydroxylation results in its oxidation into succinate, which is an end product whose accumulation can inhibit PHD activity even in the presence of oxygen [93]. In fact, deficiency of succinate dehydrogenase has been demonstrated to increase succinate levels and competitively inhibit PHDs under normoxia, leading to HIF1α stabilization in a pseudo-hypoxic phenotype [93]. Interestingly, PHD activity can be rescued by artificially increasing cellular α-ketoglutarate levels both in normoxia, reversing the succinate-mediated HIF1α stabilization [165], and hypoxia, resulting in the destabilization of HIF1α and reversing the hypoxic phenotype [166]. Therefore, PHD activity is regulated not only by oxygen availability, but also by the availability of α-ketoglutarate, a metabolite which plays a central role in numerous metabolic processes and is closely connected to amino acid metabolism [94]. In fact, both intracellular α-ketoglutarate levels and PHD activity are highly dependent on amino acid availability, while amino acids ability to induce mTORC1 signaling requires PHD enzymatic activity [167].
It is worth noting that, in addition to the principal mechanism regulating HIF1α stability in response to oxygen availability involving PHD and VHL, there are also oxygen-independent pathways regulating the synthesis and degradation of HIF1α, which involve RACK1 (receptor for activated C kinase 1) protein binding to HIF1α, recruitment of an ubiquitin ligase complex and consequent HIF1α proteasome-mediated degradation [158, 168].
HIF transcriptional targets
HIF1α and HIF2α overlap in their ability to activate target genes involved in angiogenesis, metastasis and invasion, while HIF1α alone regulates several glycolytic and apoptotic genes and HIF2α preferentially promotes the transcription of certain genes such as vascular endothelial growth factor (VEGF) or transforming growth factor α (TGFα) [155, 159, 164, 169].
HIF1 activation increases oxygen and nutrients supply to tumors through angiogenesis and erythropoiesis stimulation by VEGF and erythropoietin (EPO) upregulation, respectively [21]. In addition, as part of the molecular mechanisms associated with the Warburg effect, HIF1 enhances glycolysis and lactate production by transactivating glucose transporters and glycolytic enzymes [170]. In accordance with the increased aerobic glycolysis, HIF1 prevents the mitochondrial oxidation of pyruvate, the final product of glycolysis, by inhibiting the activity of pyruvate dehydrogenase (PDH) through pyruvate dehydrogenase kinase 1 (PDHK1) induction [171]. Furthermore, HIF1 enhances electron transport chain efficiency through cytochrome c oxidase subunit IV, isoform 2 (COX4I2) induction, which replaces the less efficient isoform 1 (COX4I1), resulting in increased ATP production and reduced ROS generation [172]. In addition to the catabolic process of anaerobic glycolysis, HIF1 also endorses anabolic processes such as glycogen synthesis by upregulating the enzymes involved in its biosynthetic pathway [173-175].
HIF1 effects on MYC
There is a complex interplay between HIF1 and MYC proteins concerning glucose metabolism and mitochondrial function [131, 156, 176]. Both HIF1 and MYC share common metabolic target genes such as SLC2A1 glucose transporter, HK, phosphofructokinase (PFK), pyruvate kinase (PK) or LDH, among others [131]. In contrast, SLC2A3 glucose transporter is a specific HIF1 target gene [131]. On the other hand, HIF1 and MYC have opposing effects on cell proliferation, mitochondrial biogenesis and DNA repair [176]. HIF1 impairs mitochondrial biogenesis and oxygen consumption by inhibiting MYC-mediated transcription and inducing MYC degradation [134], while regulates cell cycle and DNA repair genes by functionally counteracting MYC through displacement of MYC inhibitory binding from the CDKN1A promoter [177] and of MYC activating binding from MSH2 and MSH6 promoters [178]. Remarkably, HIF1 directly inhibits MYC through induction of MXI1, which binds to MAX and represses MYC transcriptional activity, and through promotion of MYC proteasomal degradation [134, 179]. Indeed, HIF increases MYC phosphorylation at Thr58, triggering MYC ubiquitination, and decreases the de-ubiquitinating enzyme USP28, promoting MYC proteasome-dependent degradation [180]. On the other hand, MYC induces MCM3 and MCM5 proteins [145] which in turn inhibit HIF1 activity by stimulating HIF1α hydroxylation, ubiquitination and degradation [181, 182]. However, prolonged hypoxic conditions reduced MCM mRNA expression in a HIF1-dependent manner, indicating that MCM and HIF1 display antagonistic functions [181, 182]. Interestingly, HIF1 and MYC present sirtuin-mediated shared regulation mechanisms since sirtuin 1 (SIRT1) works in conjunction with both transcription factors while SIRT6 inhibit their transcriptional activity via effects on chromatin [183]. Moreover, dual deficiency of oxygen and glucose suppresses HIF signaling [184] and enhances MYC degradation in cancer cells as an adaptive response to survive under conditions of deficient energy sources [185].
The PI3K/Akt pathway
Overactivation of phosphoinositide-3-kinase (PI3K)/Akt signaling is commonly observed in human cancers, as it is essential for cell proliferation, growth, survival and metabolic reprogramming [186]. PI3Ks are a family of lipid kinases that integrate prosurvival signals such as growth factors, cytokines, hormones and other environmental cues, translating them into intracellular signals that activate Akt-dependent and Akt-independent downstream signaling pathways [186]. Akt is a serine-threonine protein kinase that is mainly regulated following PI3K activation and through sequential phosphorylation at Thr308 and Ser473 [187, 188]. Since constitutive activation of Akt is frequently found in human tumors, being a central node in the PI3K/Akt signaling pathway, it is potentially interesting to molecularly target components of this pathway for cancer therapy [188].
Forkhead box O (FOXO) transcription factors are direct targets of Akt that modulate cell cycle, growth, DNA repair, survival, apoptosis, metabolism, cellular differentiation, resistance to oxidative stress and tumor suppressor pathways [189-193]. Four different FOXO proteins are encoded in mammalian cells; FOXO1, FOXO3a and FOXO4, which are ubiquitously expressed, and FOXO6, which is expressed predominantly in neural cells [194, 195]. Post-translational modifications such as phosphorylation, acetylation and ubiquitination regulate the translocation of FOXO proteins to the nucleus [196] where they activate transcription by binding to gene regulatory regions [197]. The principal mechanism of FOXO transcriptional regulation is FOXO phosphorylation by Akt which impairs its DNA binding activity and promotes its interaction with the chaperone protein 14-3-3. This interaction triggers the nuclear exclusion, cytoplasmic accumulation and ubiquitin-proteasome pathway-dependent degradation of FOXO factors, and promotes cell survival [198, 199]. In the presence of oxidative stress, FOXO proteins are activated and released from 14-3-3 through Jun N-terminal kinase (JNK) signaling [189, 191, 200, 201] (Figure 5).
FOXM1 transcription factor is a crucial regulator of cell proliferation and cell cycle progression that is overexpressed in many types of cancer. Cell differentiation, angiogenesis, senescence, DNA damage repair and tissue homeostasis are regulated by FOXM1, conferring oncogene-like properties to this forkhead subfamily member [202]. Recent studies have reported that FOXO3a represses FOXM1 expression and that they both compete for binding to similar DNA sequences, sharing numerous target genes but being antagonists [196, 203-205]. It is worth noting that FOXO3a and FOXM1 proteins are indirect targets of several conventional and widely used cytotoxic chemotherapeutic drugs such as cisplatin or gefitinib, which mediate their effects through FOXO3a activation and FOXM1 indirect repression via PI3K/Akt signaling pathway inhibition [206-211]. The dysregulation of the PI3K/Akt/FOXO3a axis may lead to drug resistance by enhancing DNA repair, as well as cancer cell maintenance, proliferation and survival through overexpression of FOXM1 [196, 202].
A hallmark of most cancers where the PI3K/Akt pathway is hyperactivated is the inactivation of FOXO proteins [188, 201], postulating FOXO family members as tumor suppressors [212]. Accordingly, PI3K depletion results in FOXO proteins activation, induction of apoptosis, decrease of cell viability and G1 cell cycle arrest with inhibition of CDK4/6, cyclin D and accumulation of p27Kip1 [213]. Indeed, in vivo models of loss of FOXO function exhibit spontaneous tumor formation, while FOXO overexpression can inhibit tumorigenesis [191, 211, 212, 214-216]. Even though FOXO transcription factors are considered to be tumor suppressors, genetic inactivation of FOXO is not often found in human cancers, being predominantly repressed through overactivation of the PI3K/Akt pathway caused by mutations in RAS, PTEN or PI3K genes [212]. Therefore, the search for compounds that promote activation and relocalisation of FOXO from the cytoplasm to the nucleus is a promising therapeutic approach for cancer treatment and overcoming drug resistance [215, 217].
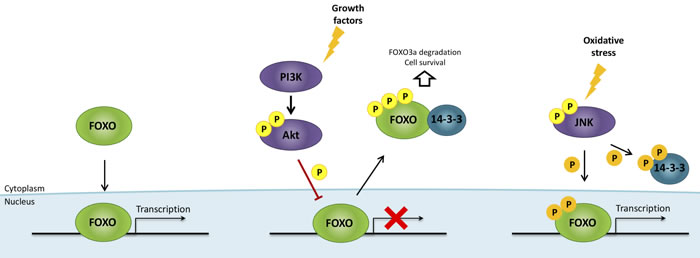
Figure 5: FOXO regulation by growth factors and oxidative stress. Growth factors activate PI3K/Akt pathway, resulting in FOXO factors phosphorylation, impairment of FOXO binding activity to DNA and promotion of FOXO interaction with the chaperone protein 14-3-3, which in turn causes FOXO nuclear exclusion, cytoplasmic accumulation and ubiquitin-proteasome pathway-dependent degradation. Oxidative stress activates Jun N-terminal kinase (JNK) signaling, which phosphorylates both FOXO (at other regulatory sites than Akt) and 14-3-3 proteins, triggering the release of FOXO factors, their nuclear translocation and their transcriptional activity. Akt/PBK, protein kinase B; FOXO, forkhead box O; P, phosphate; PI3K, phosphoinositide-3-kinase.
FOXO effects on MYC, HIF1α and mTOR
Activated PI3K/Akt pathway stimulates cell growth and proliferation, and stabilizes MYC through inhibition of GSK3β by preventing MYC phosphorylation at Thr58 [154]. Active PI3K and MYC specifically cooperate in dysregulation of cell growth and proliferation, since both regulate a common set of cellular processes [218]. Conversely, activation of FOXO transcription factors following inhibition of PI3K/Akt signaling represses multiple MYC target genes including those involved in cell proliferation and mitochondrial activity, blocking MYC-mediated cell proliferation and transformation, and reducing ROS production [219, 220]. In addition, FOXO3a induces the expression of the MAD/MXD family of transcriptional repressors, although MXI1 is the only member that is its direct target. Indeed, MXI1 is necessary for efficient inhibition of MYC transcriptional activity [221]. Furthermore, FOXO3a activation considerably reduces MYC protein levels by enhancing phosphorylation of MYC at Thr58, which triggers its proteasomal degradation [220]. Interestingly, FOXO3a-mediated regulation of MYC at different levels enables both acute inhibition of mitochondrial gene expression by MYC degradation and sustained inhibition through MXI1 antagonistic effects [222]. Therefore, the inhibition of the transcriptional activity of FOXO proteins by Akt-mediated phosphorylation is required for MYC-induced cell proliferation and transformation [219].
FOXO3a is induced under hypoxic conditions as a direct target gene of HIF1 to mediate the hypoxic repression of nuclear-encoded genes with mitochondrial function by directly antagonizing MYC at their promoters, resulting in reduced mitochondrial mass, oxygen consumption and ROS production [223, 224]. Additionally, FOXO3a promotes cell survival both in hypoxic tumor cells and hypoxic tumor tissue in vivo, in contrast with its role as a tumor suppressor under normoxic conditions [212, 223]. On the other hand, FOXO3a prevents HIF1α stabilization by blocking the hypoxia-induced ROS increase [220], and inhibits HIF1α activity through stimulation of CITED2 (Cbp/p300-interacting transactivator 2) expression, also reducing HIF1α-induced apoptosis during hypoxic stress and promoting cell survival [224].
In response to energy stress, FOXO proteins inhibit the mechanistic target of rapamycin complex 1 (mTORC1) signaling through induction of BCL2/adenovirus E1B 19kDa interacting protein 3 (BNIP3) expression, which in turn negatively regulates the mTORC1 activator RHEB and the BCL2 pro-survival family members, resulting in energy stress-induced apoptosis [225]. In addition, mTORC1 inhibition upregulates FOXO3a expression and nuclear accumulation through FOXO3a demethylation [226]. Conversely, mTOR complex 2 (mTORC2) phosphorylates the Class IIa histone deacetylases (HDACs) in an Akt-independent manner, resulting in FOXO acetylation, release of MYC proteins from FOXO-mediated repression and the consequent conferral of resistance to PI3K and Akt Inhibitors [227].
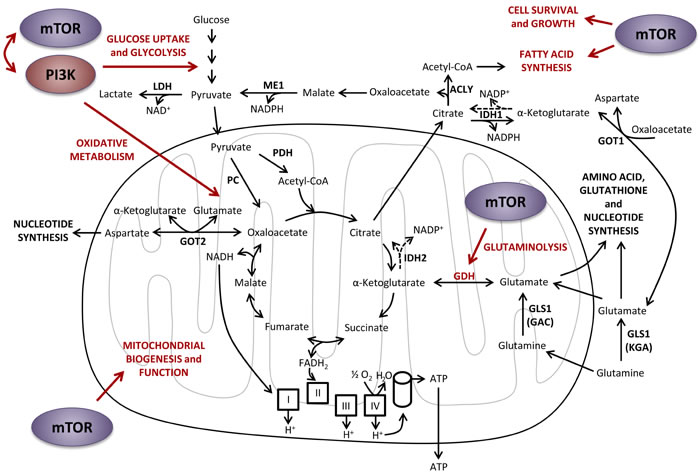
Figure 6: Effects of PI3K and mTOR on central carbon metabolism. PI3K and mTOR signaling pathways positively regulate each other’s activity, as well as glucose uptake and glycolysis, oxidative metabolism and glutaminolysis. mTOR also promotes fatty acids synthesis and cell survival and growth. ACLY, ATP citrate lyase; ATP, adenosine triphosphate; CoA, coenzyme A; FADH2, flavin adenine dinucleotide reduced form; GAC, glutaminase C; GDH, glutamate dehydrogenase; GLS1, glutaminase 1; GOT1, glutamic-oxaloacetic transaminase 1 cytoplasmic form; GOT2, glutamic-oxaloacetic transaminase 2 mitochondrial form; IDH1, isocitrate dehydrogenase cytoplasmic form; IDH2, isocitrate dehydrogenase mitochondrial form; KGA, kidney (K-type) glutaminase; LDH, lactate dehydrogenase; ME1, malic enzyme 1 cytoplasmic form; NAD+, nicotinamide adenine dinucleotide oxidized form; NADH, nicotinamide adenine dinucleotide reduced form; NADP+, nicotinamide adenine dinucleotide phosphate oxidized form; NADPH, nicotinamide adenine dinucleotide phosphate reduced form; PC, pyruvate carboxylase; PDH, pyruvate dehydrogenase.
mTOR
The mechanistic target of rapamycin (mTOR, formerly mammalian TOR) is a conserved cytoplasmic serine-threonine protein kinase that acts as a central cell growth regulator by sensing mitogens, energy and amino acids. mTOR pathway regulates cell survival and growth through modulation of some pivotal cellular processes including protein synthesis, ribosome biogenesis, autophagy and metabolism [228]. In fact, the dysregulation of mTOR-dependent cellular homeostasis maintenance is associated with several human diseases such as cancer and considerable research efforts have been made to efficiently inhibit mTOR signaling [229, 230].
mTOR forms two functionally and structurally different multiprotein complexes named mTOR complex 1 (mTORC1) and 2 (mTORC2). mTORC1 activity is regulated by growth factors, oxygen and nutrient availability. Activation of PI3K/Akt and RAS/RAF/ERK pathways by growth factors results in Akt- and ERK-mediated phosphorylation and inactivation of the heterodimer tuberous sclerosis 1 (TSC1)/TSC2, which is a GTPase-activating protein (GAP) that negatively regulates mTORC1 through inhibition of the RAS homolog enriched in brain (RHEB) GTPase [231]. Remarkably, intracellular amino acids are necessary for the activation of mTORC1 since they activate the mechanism by which mTORC1 is able to interact with and be activated by RHEB [67].
Activated mTORC1 signaling cascade initiates with the direct phosphorylation of the regulators of translation eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1) and S6 kinase 1 (S6K1), which promote protein synthesis [232]. In addition, mTORC1 regulates lipid homeostasis through activation of the transcription factors sterol regulatory element-binding protein 1/2 (SREBP1/2), which in turn control the expression of genes involved in fatty acid, triglyceride, phospholipid and cholesterol synthesis [228, 233]. Interestingly, mTORC1 also promotes mitochondrial biogenesis and the expression of genes involved in oxidative metabolism [234, 235]. On the other hand, mTORC2 pathway regulation and function remain poorly understood. mTORC2 signaling is independent of nutrient availability but is sensitive to PI3K signaling [228]. mTORC2 directly activates Akt through phosphorylation of the Ser473 residue, which in turn activates mTORC1, both situating mTOR upstream and downstream of Akt [236]. It is worth noting that acute rapamycin treatment specifically inhibits mTOR when it is part of mTORC1 but not of mTORC2 [228, 229].
mTOR regulation by hypoxia and MYC
Hypoxic oxygen levels inhibit mTORC1 by activating the TSC1/TSC2 complex through two different pathways. On the one hand, hypoxia reduces cellular ATP levels and triggers 5’-AMP-activated protein kinase (AMPK) activation, which positively regulates TSC1/TSC2 in a HIF1-independent manner [237]. On the other hand, hypoxia activates TSC1/TSC2 by the transcriptional induction of regulated in development and DNA damage responses 1 (REDD1) gene, antagonizing other pathways that promote growth through TSC1/TSC2 inhibition via Akt [238, 239]. Hypoxia can also negatively regulate mTORC1 through the hypoxia-inducible protein BNIP3 binding to RHEB, which inhibits the ability of RHEB to activate mTORC1 [240]. Conversely, MYC acts as a strong and direct repressor for TSC2 expression by binding to its promoter, resulting in mTORC1 activation [241]. In addition, mTORC1 downstream effector S6K1 phosphorylates the eukaryotic initiation factor eIF4B, enhancing MYC translation efficiency and positively regulating glutaminase (GLS) and glutamate dehydrogenase (GDH) [101, 242]. Moreover, glutaminolysis and α-ketoglutarate production, in response to glutamine and leucine [68], also mediate mTORC1 activation [111].
In summary, mTOR, PI3K, HIF and MYC are key regulators of cellular metabolism that are frequently altered in cancer, collaborating in both synergistic and antagonistic ways. A better understanding of the relationship between these pathways as well as the identification of other key players in the regulation of the tumor metabolic reprogramming are fundamental challenges for the development of new strategies for cancer treatment.
Therapeutic perspectives
Targeting metabolic reprogramming in cancer therapy
Development of malignancy is accompanied by a complete metabolic reprogramming closely related to the acquisition of most of cancer hallmarks [2, 28]. Many known genetic and epigenetic alterations converge in a common adaptation of tumor cell metabolism [30]. Indeed, metabolic properties of tumor cells are significantly different from those of non-transformed cells. In addition, tumor metabolic reprogramming is linked to drug resistance in cancer treatment [243, 244]. Accordingly, metabolic adaptations are also involved in different therapeutic approaches for cancer therapy. It is worth noting that some of the first chemotherapeutical agents used in cancer treatment were antimetabolites, such as aminopterin, methotrexate or 5-fluorouracil, that impaired the nucleotide synthesis and DNA replication [245, 246]. From then on, numerous metabolic pathways and enzymes have been successfully tested as anticancer targets [247]. Since aerobic glycolysis is one of the key metabolic features of cancer cells, many studies are focused on inhibiting this pathway by blocking the enzymes that control it [248]. Targeting the PPP with dehydroepiandrosterone (DHEA) and oxythiamine to respectively inhibit G6PD and TKT has also proven to have antitumor effects [50, 244, 249]. Interestingly, promoting pyruvate dehydrogenase (PDH) activity with dichloroacetate (DCA) presents promising results with minor side effects in early phase clinical trials with glioblastoma patients by suppressing angiogenesis, increasing mitochondrial ROS, inducing apoptosis, blocking HIF1 signaling and activating tumor suppressor p53 [250-252]. In fact, DCA inhibits PDHK leading to the metabolic switch from glycolysis to oxidative phosphorylation through PDH reactivation [250]. Moreover, combined therapies with DCA and conventional cancer therapeutics such as omeprazole and tamoxifen show synergistic antitumor effects which can overcome drug resistance [253]. Ongoing clinical trials with DCA as a single agent or in combination with other therapeutics are being conducted for patients with recurrent or metastatic solid tumors and head and neck carcinoma (clinical trials NCT00566410 and NCT01386632).
There is a growing interest on the development of pharmacological strategies to inhibit tumor glutamine metabolism. The use of amino acid analogues such as acivicin led to severe side effects in clinical trials, aiming for more selective therapeutic strategies [254]. As a result, GLS1 isoform has emerged as a promising target for cancer therapy and several specific small molecule inhibitors of GLS1 have recently been characterized. Compound 968 and BPTES (bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide) are two allosteric inhibitors of GLS1 that exhibit antitumor activities in numerous pre-clinical studies and several tumor types [100, 108, 121, 255-257]. Remarkably, the selective inhibitor of GLS1 known as CB-839 presents in vitro antiproliferative activity against acute myeloid leukemia cells [258], and a panel of triple-negative breast cancer cell lines, but not estrogen receptor (ER) or human epidermal growth factor receptor 2 (HER2) positive cell lines, as well as in vivo efficacy in breast cancer xenograft models [107]. In addition, CB-839 has recently entered phase I clinical trials without displaying central nervous system toxicity (clinical trials NCT02071862 and NCT02071888). In fact, GLS1 inhibition is a good strategy for tumor cells that overexpress MYC and thus present glutamine dependence [22, 99]. It is worth mentioning that, to date, no effective MYC inhibitors have been developed despite the fact that MYC overexpression is frequently found in human cancers [125, 259-261]. However, targeting GLS1 significantly antagonizes the growth of tumors presenting MYC overexpression and can be exploited as a novel antitumor therapy [100].
Combination therapies
The identification of cytotoxic compounds has led the development of antitumor therapeutics until in the recent years chemotherapy advanced into the era of molecularly targeted therapeutics [262]. The bases of molecular targeted cancer therapy are to selectively kill tumor cells while sparing non-malignant cells, and prevent tumor resistance emergence and relapse [263]. However, solid tumors response to targeted monotherapy is limited and frequently associated with the development of drug resistance. In addition, the design of targeted therapies requires the definition of the activated oncogenic pathways in transformed cells and the availability of selective small-molecule inhibitors directed to these pathways. The modest efficacy of current therapies is also caused by the high degree of tumor clonal and genetic heterogeneity, since inhibition of a single target does not necessarily eradicate the tumor. Therefore, the use of combination therapies of selective agents and/or cytotoxic agents that inhibit two or more molecular targets in a single pathway, or in parallel or compensatory pathways, is an attractive strategy for cancer treatment [264]. Additionally, the simultaneous inhibition of multiple targets or redundant pathways is aimed at improving treatment efficacy and overcoming and/or preventing the emergence of adaptive resistance.
Then, in order to select appropriate molecular targets for inhibition or modification, is necessary to first perform a tumor expression profiling to identify its specific oncogenic signatures, and confirm that the target is tumor specific, non-redundant, and able to influence the outcome of tumor progression [263, 265]. However, many oncogenic pathways cannot be directly targeted with small-molecule inhibitors [260]. Remarkably, gene expression analysis can be used as a predictive tool to identify the oncogenic pathways which are dysregulated in a specific tumor, providing a potential basis for guiding the use of pathway-specific drugs and directing combination therapies aimed to slow tumor growth and progression, improve treatment response, and overcome therapeutic resistance [265].
Concluding remarks
The understanding of cancer cell biology is of pivotal importance to identify biomarkers for early diagnosis and design new therapeutic strategies. In particular, tumor cells switch their core metabolism to meet the increased requirements of cell growth and division. Accordingly, oncogenic signals converge to reprogram tumor metabolism by enhancing key metabolic pathways such as glycolysis, PPP, glutaminolysis and amino acid, lipid and nucleic acid metabolism [19, 63]. Therefore, tumor metabolic reprogramming is a direct result of the re-engineering of intracellular signaling pathways that are altered by activating mutations in oncogenes, loss-of-function mutations in tumor suppressor genes and epigenetic modifications, which finally gives to transformed cells a proliferative advantage over non-malignant cells [5, 6]. However, the final tumor phenotype depends on the homeostatic nature of metabolism, since metabolic rewiring is associated with compensatory and regulatory adjustments. There are several levels of metabolic adaptations, from changes in the concentrations and fluxes associated with substrate-enzyme affinities to adjustments coordinated by metabolite-based regulatory loops or determined by changes in the activity of regulatory proteins (e.g. MYC and HIF). In fact, metabolic rewiring allows tumor cells to exhibit rapidly adaptive responses to changes in tumor microenvironment, promoting tumor progression and acquired resistance to targeted therapeutics [266, 267]. On the other hand, tumor metabolic adaptation after single-agent treatment can reveal new cell dependences and vulnerabilities which, in turn, may be potential candidates to be targeted in combination therapies. The final aim of the combination treatments is to achieve synergistic therapeutic effect, dose and toxicity reduction, and to minimize or delay the induction of drug resistance [268].
Personalized medicine in cancer requires the correct diagnosis of cancer to give patients the most appropriate treatment according to their individual circumstances and the molecular characteristics of their tumors. However, personalized medicine is still at a relatively early stage in its development, and therefore the classification of cancers is based on critical molecular targets identified by translational research. Hence, more efforts should be put into understanding the tumor biology in order to identify the involved targets and determine the optimum treatment for each specific tumor. Likewise, the developing of new therapeutic strategies that specifically target the molecular pathways involved in promoting tumor cell proliferation and survival, such as targeted therapies, is a major focus of cancer research today [263]. However, the currently available chemotherapeutic treatments exhibit modest efficacy due to their side effects and drug resistance [267]. In addition, the design of more efficient targeted therapies requires a better definition of the activated oncogenic pathways in transformed cells and the availability of selective small-molecule inhibitors directed to these pathways. In this context, the search for combined chemotherapies with low systemic toxicity that inhibit two or more molecular targets in a single pathway, or in redundant or compensatory pathways, is a promising strategy for cancer treatment [264]. With this purpose, both metabolic tumor characterization and gene expression analysis can be used to identify the dysregulated molecular pathways in a specific tumor, providing a potential basis for guiding the use of target-specific drugs and directing combination therapies [265].
Acknowledgments
The authors’ laboratory receives financial support from the Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR)-Generalitat de Catalunya (2014SGR1017) and the Ministerio de Economía y Competitividad from the Spanish Government and FEDER funds from the European Union “una manera de hacer Europa” (SAF2014-56059-R). MC acknowledges the support received through the prize ICREA Academia for excellence in research, funded by ICREA Foundation-Generalitat de Catalunya.
Conflicts of interest
The authors declare no conflict of interest.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015; 136:E359-386.
2. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646-674.
3. Shortt J and Johnstone RW. Oncogenes in cell survival and cell death. Cold Spring Harb Perspect Biol. 2012; 4.
4. Hanahan D and Weinberg RA. The hallmarks of cancer. Cell. 2000; 100:57-70.
5. Qiu B and Simon MC. Oncogenes strike a balance between cellular growth and homeostasis. Semin Cell Dev Biol. 2015.
6. Morris LG and Chan TA. Therapeutic targeting of tumor suppressor genes. Cancer. 2015; 121:1357-1368.
7. Lee J-K, Choi Y-L, Kwon M and Park PJ. Mechanisms and Consequences of Cancer Genome Instability: Lessons from Genome Sequencing Studies. Annual Review of Pathology: Mechanisms of Disease. 2015; 11.
8. Negrini S, Gorgoulis VG and Halazonetis TD. Genomic instability — an evolving hallmark of cancer. Nature Reviews Molecular Cell Biology. 2010; 11:220-228.
9. Venkatesan S, Natarajan AT and Hande MP. Chromosomal instability—mechanisms and consequences. Mutat Res Genet Toxicol Environ Mutagen. 2015; 793:176-84.(doi):10.1016/j.mrgentox.2015.1008.1008. Epub 2015 Aug 1028.
10. Yen C-Y, Huang H-W, Shu C-W, Hou M-F, Yuan S-SF, Wang H-R, Chang Y-T, Farooqi AA, Tang J-Y and Chang H-W. DNA methylation, histone acetylation and methylation of epigenetic modifications as a therapeutic approach for cancers. Cancer Letters. 2016; 373:185-192.
11. Feinberg AP and Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004; 4:143-153.
12. Gokul G and Khosla S. DNA methylation and cancer. Subcell Biochem. 2013; 61:597-625.
13. Wang X and Jin H. The epigenetic basis of the Warburg effect. Epigenetics. 2014; 5:566-568.
14. Liu X, Wang X, Zhang J, Lam EKY, Shin VY, Cheng ASL, Yu J, Chan FKL, Sung JJY and Jin HC. Warburg effect revisited: an epigenetic link between glycolysis and gastric carcinogenesis. Oncogene. 2009; 29:442-450.
15. Nordgren KK and Skildum AJ. The deep end of the metabolite pool: influences on epigenetic regulatory mechanisms in cancer. European Journal of Clinical Investigation. 2015; 45:9-15.
16. Guan KL and Xiong Y. Regulation of intermediary metabolism by protein acetylation. Trends Biochem Sci. 2011; 36:108-116.
17. Wu Q and Ni X. ROS-Mediated DNA Methylation Pattern Alterations in Carcinogenesis. Current Drug Targets. 2015; 16:13-19.
18. DeBerardinis RJ, Lum JJ, Hatzivassiliou G and Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell metabolism. 2008; 7:11-20.
19. Ward PS and Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer cell. 2012; 21:297-308.
20. Cairns RA, Harris IS and Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011; 11:85-95.
21. Courtnay R, Ngo DC, Malik N, Ververis K, Tortorella SM and Karagiannis TC. Cancer metabolism and the Warburg effect: the role of HIF-1 and PI3K. Mol Biol Rep. 2015; 42:841-851.
22. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB and Thompson CB. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105:18782-18787.
23. DeBerardinis RJ, Sayed N, Ditsworth D and Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008; 18:54-61.
24. Warburg O, Posener K and Negelein E. Über den Stoffwechsel der Karzinomzellen. Biochemische Zeitschrift. 1924; 152:309-344.
25. Warburg O. On the origin of cancer cells. Science (New York, NY. 1956; 123:309-314.
26. Semenza GL, Artemov D, Bedi A, Bhujwalla Z, Chiles K, Feldser D, Laughner E, Ravi R, Simons J, Taghavi P and Zhong H. ‘The metabolism of tumours’: 70 years later. Novartis Found Symp. 2001; 240:251-260; discussion 260-254.
27. Gogvadze V, Zhivotovsky B and Orrenius S. The Warburg effect and mitochondrial stability in cancer cells. Mol Aspects Med. 2010; 31:60-74.
28. Kroemer G and Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer cell. 2008; 13:472-482.
29. Salway JG. (2000). Metabolism at a Glance. (Oxford: Blackwell Science).
30. Vander Heiden MG, Cantley LC and Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (New York, NY. 2009; 324:1029-1033.
31. Som P, Atkins HL, Bandoypadhyay D, Fowler JS, MacGregor RR, Matsui K, Oster ZH, Sacker DF, Shiue CY, Turner H, Wan CN, Wolf AP and Zabinski SV. A fluorinated glucose analog, 2-fluoro-2-deoxy-D-glucose (F-18): nontoxic tracer for rapid tumor detection. J Nucl Med. 1980; 21:670-675.
32. Bayley JP and Devilee P. The Warburg effect in 2012. Curr Opin Oncol. 2012; 24:62-67.
33. Yeung SJ, Pan J and Lee MH. Roles of p53, MYC and HIF-1 in regulating glycolysis - the seventh hallmark of cancer. Cell Mol Life Sci. 2008; 65:3981-3999.
34. Li J, Zhu S, Tong J, Hao H, Yang J, Liu Z and Wang Y. Suppression of lactate dehydrogenase A compromises tumor progression by downregulation of the Warburg effect in glioblastoma. Neuroreport. 2016; 27:110-115.
35. Liberti MV and Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci. 2016; 41:211-218.
36. Yamamoto T, Seino Y, Fukumoto H, Koh G, Yano H, Inagaki N, Yamada Y, Inoue K, Manabe T and Imura H. Over-expression of facilitative glucose transporter genes in human cancer. Biochemical and biophysical research communications. 1990; 170:223-230.
37. Altenberg B and Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004; 84:1014-1020.
38. Levine AJ and Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science (New York, NY. 2010; 330:1340-1344.
39. Cerella C, Dicato M and Diederich M. Modulatory roles of glycolytic enzymes in cell death. Biochem Pharmacol. 2014; 92:22-30.
40. Kim JW and Dang CV. Multifaceted roles of glycolytic enzymes. Trends Biochem Sci. 2005; 30:142-150.
41. Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L, Guo X, Chang LJ, You MJ, Koochekpour S, Saleem M, Huang H, Lu J, et al. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014; 8:1461-1474.
42. Anderson M, Marayati R, Moffitt R and Jen Yeh J. Hexokinase 2 promotes tumor growth and metastasis by regulating lactate production in pancreatic cancer. Oncotarget. 2016; doi:10.18632/oncotarget.9760.
43. Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R, Hawkins C and Guha A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med. 2011; 208:313-326.
44. Pastorino JG, Shulga N and Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. The Journal of biological chemistry. 2002; 277:7610-7618.
45. Roberts DJ and Miyamoto S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015; 22:248-257.
46. Patra KC and Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. 2014; 39:347-354.
47. Li D, Zhu Y, Tang Q, Lu H, Li H, Yang Y, Li Z and Tong S. A new G6PD knockdown tumor-cell line with reduced proliferation and increased susceptibility to oxidative stress. Cancer Biother Radiopharm. 2009; 24:81-90.
48. Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M and Yang X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. 2011; 13:310-316.
49. Qiu Z, Guo W, Wang Q, Chen Z, Huang S, Zhao F, Yao M, Zhao Y and He X. MicroRNA-124 Reduces Pentose Phosphate Pathway and Proliferation by Targeting PRPS1 and RPIA mRNAs in Human Colorectal Cancer Cells. Gastroenterology. 2015.
50. Boros LG, Puigjaner J, Cascante M, Lee WN, Brandes JL, Bassilian S, Yusuf FI, Williams RD, Muscarella P, Melvin WS and Schirmer WJ. Oxythiamine and dehydroepiandrosterone inhibit the nonoxidative synthesis of ribose and tumor cell proliferation. Cancer Res. 1997; 57:4242-4248.
51. Boren J, Montoya AR, de Atauri P, Comin-Anduix B, Cortes A, Centelles JJ, Frederiks WM, Van Noorden CJ and Cascante M. Metabolic control analysis aimed at the ribose synthesis pathways of tumor cells: a new strategy for antitumor drug development. Mol Biol Rep. 2002; 29:7-12.
52. Comin-Anduix B, Boren J, Martinez S, Moro C, Centelles JJ, Trebukhina R, Petushok N, Lee WN, Boros LG and Cascante M. The effect of thiamine supplementation on tumour proliferation. A metabolic control analysis study. European journal of biochemistry / FEBS. 2001; 268:4177-4182.
53. Vizan P, Alcarraz-Vizan G, Diaz-Moralli S, Solovjeva ON, Frederiks WM and Cascante M. Modulation of pentose phosphate pathway during cell cycle progression in human colon adenocarcinoma cell line HT29. Int J Cancer. 2009; 124:2789-2796.
54. Tudzarova S, Colombo SL, Stoeber K, Carcamo S, Williams GH and Moncada S. Two ubiquitin ligases, APC/C-Cdh1 and SKP1-CUL1-F (SCF)-beta-TrCP, sequentially regulate glycolysis during the cell cycle. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108:5278-5283.
55. Cardozo T and Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004; 5:739-751.
56. Santos CR and Schulze A. Lipid metabolism in cancer. FEBS J. 2012; 279:2610-2623.
57. Huang C and Freter C. Lipid metabolism, apoptosis and cancer therapy. Int J Mol Sci. 2015; 16:924-949.
58. Swinnen JV, Brusselmans K and Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care. 2006; 9:358-365.
59. Yahagi N, Shimano H, Hasegawa K, Ohashi K, Matsuzaka T, Najima Y, Sekiya M, Tomita S, Okazaki H, Tamura Y, Iizuka Y, Nagai R, Ishibashi S, et al. Co-ordinate activation of lipogenic enzymes in hepatocellular carcinoma. Eur J Cancer. 2005; 41:1316-1322.
60. Milgraum LZ, Witters LA, Pasternack GR and Kuhajda FP. Enzymes of the fatty acid synthesis pathway are highly expressed in in situ breast carcinoma. Clin Cancer Res. 1997; 3:2115-2120.
61. Zhang F and Du G. Dysregulated lipid metabolism in cancer. World J Biol Chem. 2012; 3:167-174.
62. Icard P, Poulain L and Lincet H. Understanding the central role of citrate in the metabolism of cancer cells. Biochimica et biophysica acta. 2012; 1825:111-116.
63. Phan LM, Yeung SC and Lee MH. Cancer metabolic reprogramming: importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol Med. 2014; 11:1-19.
64. Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, Downward J and Schulze A. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene. 2005; 24:6465-6481.
65. Berwick DC, Hers I, Heesom KJ, Moule SK and Tavare JM. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. The Journal of biological chemistry. 2002; 277:33895-33900.
66. Menendez JA and Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007; 7:763-777.
67. Yuan HX, Xiong Y and Guan KL. Nutrient sensing, metabolism, and cell growth control. Molecular cell. 2013; 49:379-387.
68. Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, MacKeigan JP, Porter JA, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009; 136:521-534.
69. Lamb RF. Amino acid sensing mechanisms: an Achilles heel in cancer? FEBS J. 2012; 279:2624-2631.
70. Liu W, Le A, Hancock C, Lane AN, Dang CV, Fan TW and Phang JM. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109:8983-8988.
71. Tsun ZY and Possemato R. Amino acid management in cancer. Semin Cell Dev Biol. 2015.
72. Bhutia YD, Babu E, Ramachandran S and Ganapathy V. Amino Acid transporters in cancer and their relevance to “glutamine addiction”: novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015; 75:1782-1788.
73. Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT and Dang CV. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009; 458:762-765.
74. Kim J, Lee JH and Iyer VR. Global identification of Myc target genes reveals its direct role in mitochondrial biogenesis and its E-box usage in vivo. PloS one. 2008; 3:e1798.
75. Zhu M, Wang N, Tsao SW, Yuen MF, Feng Y, Wan TS and Man K. Up-regulation of microRNAs, miR21 and miR23a in human liver cancer cells treated with Coptidis rhizoma aqueous extract. Exp Ther Med. 2011; 2:27-32.
76. Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013; 13:572-583.
77. Tedeschi PM, Markert EK, Gounder M, Lin H, Dvorzhinski D, Dolfi SC, Chan LL, Qiu J, DiPaola RS, Hirshfield KM, Boros LG, Bertino JR, Oltvai ZN, et al. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell Death Dis. 2013; 4:e877.
78. Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB and Mootha VK. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science (New York, NY. 2012; 336:1040-1044.
79. di Salvo ML, Contestabile R, Paiardini A and Maras B. Glycine consumption and mitochondrial serine hydroxymethyltransferase in cancer cells: the heme connection. Med Hypotheses. 2013; 80:633-636.
80. Phang JM, Liu W, Hancock CN and Fischer JW. Proline metabolism and cancer: emerging links to glutamine and collagen. Curr Opin Clin Nutr Metab Care. 2015; 18:71-77.
81. Liu W and Phang JM. Proline dehydrogenase (oxidase) in cancer. Biofactors. 2012; 38:398-406.
82. Daye D and Wellen KE. Metabolic reprogramming in cancer: unraveling the role of glutamine in tumorigenesis. Semin Cell Dev Biol. 2012; 23:362-369.
83. Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012; 12:685-698.
84. Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR and Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107:8788-8793.
85. Dang CV. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med. 2013; 3.
86. Gaude E and Frezza C. Defects in mitochondrial metabolism and cancer. Cancer Metab. 2014; 2:10.
87. Acin-Perez R and Enriquez JA. The function of the respiratory supercomplexes: the plasticity model. Biochimica et biophysica acta. 2014; 1837:444-450.
88. Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M, Su SM, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer cell. 2010; 17:225-234.
89. Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Seo SI, Lee JY, Yoo NJ and Lee SH. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer. 2009; 125:353-355.
90. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012; 26:1326-1338.
91. Desideri E, Vegliante R and Ciriolo MR. Mitochondrial dysfunctions in cancer: genetic defects and oncogenic signaling impinging on TCA cycle activity. Cancer letters. 2015; 356:217-223.
92. Chen J-Q and Russo J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 2012; 1826:370-384.
93. Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB and Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer cell. 2005; 7:77-85.
94. Boulahbel H, Duran RV and Gottlieb E. Prolyl hydroxylases as regulators of cell metabolism. Biochem Soc Trans. 2009; 37:291-294. doi: 210.1042/BST0370291.
95. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer cell. 2011; 19:17-30.
96. Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, Ding J, Lei Q, Guan KL, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science (New York, NY. 2009; 324:261-265.
97. Losman JA and Kaelin WG, Jr. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013; 27:836-852.
98. DeBerardinis RJ and Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010; 29:313-324.
99. Wise DR and Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010; 35:427-433.
100. Yuneva MO, Fan TW, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, Mates JM, Alonso FJ, Wang C, Seo Y, Chen X and Bishop JM. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell metabolism. 2012; 15:157-170.
101. Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, Jeong SM, Dempsey JM, Parkhitko A, Morrison T, Henske EP, Haigis MC, Cantley LC, et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell. 2013; 153:840-854.
102. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013; 496:101-105.
103. Jeong SM, Xiao C, Finley LW, Lahusen T, Souza AL, Pierce K, Li YH, Wang X, Laurent G, German NJ, Xu X, Li C, Wang RH, et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer cell. 2013; 23:450-463.
104. Hu W, Zhang C, Wu R, Sun Y, Levine A and Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107:7455-7460.
105. Kim MH and Kim H. Oncogenes and tumor suppressors regulate glutamine metabolism in cancer cells. J Cancer Prev. 2013; 18:221-226.
106. Iacobazzi V and Infantino V. Citrate—new functions for an old metabolite. Biol Chem. 2014; 395:387-399.
107. Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, Mackinnon AL, Parlati F, Rodriguez ML, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. 2014; 13:890-901.
108. Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, Zimmerman LJ, Liebler DC, Slebos RJ, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell metabolism. 2012; 15:110-121.
109. Thangavelu K, Chong QY, Low BC and Sivaraman J. Structural basis for the active site inhibition mechanism of human kidney-type glutaminase (KGA). Sci Rep. 2014; 4:3827.
110. Mates JM, Segura JA, Martin-Rufian M, Campos-Sandoval JA, Alonso FJ and Marquez J. Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr Mol Med. 2013; 13:514-534.
111. Duran RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E and Hall MN. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell. 2012; 47:349-358.
112. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S and Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104:19345-19350.
113. Zu XL and Guppy M. Cancer metabolism: facts, fantasy, and fiction. Biochemical and biophysical research communications. 2004; 313:459-465.
114. Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006; 6:813-823.
115. Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T and Rabinowitz JD. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol. 2013; 9:712.
116. Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG and DeBerardinis RJ. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009; 69:7986-7993.
117. Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Mates JM and DeBerardinis RJ. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108:8674-8679.
118. Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, Kelleher JK, Vander Heiden MG, Iliopoulos O, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012; 481:380-384.
119. Fendt SM, Bell EL, Keibler MA, Olenchock BA, Mayers JR, Wasylenko TM, Vokes NI, Guarente L, Vander Heiden MG and Stephanopoulos G. Reductive glutamine metabolism is a function of the alpha-ketoglutarate to citrate ratio in cells. Nat Commun. 2013; 4:2236.
120. Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS and DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012; 481:385-388.
121. Gameiro PA, Yang J, Metelo AM, Perez-Carro R, Baker R, Wang Z, Arreola A, Rathmell WK, Olumi A, Lopez-Larrubia P, Stephanopoulos G and Iliopoulos O. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell metabolism. 2013; 17:372-385.
122. Meyer N and Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008; 8:976-990.
123. Gabay M, Li Y and Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med. 2014; 4.
124. Patel JH, Loboda AP, Showe MK, Showe LC and McMahon SB. Analysis of genomic targets reveals complex functions of MYC. Nat Rev Cancer. 2004; 4:562-568.
125. Wahlstrom T and Arsenian Henriksson M. Impact of MYC in regulation of tumor cell metabolism. Biochimica et biophysica acta. 2015; 1849:563-569.
126. Adhikary S and Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol. 2005; 6:635-645.
127. Herkert B and Eilers M. Transcriptional repression: the dark side of myc. Genes Cancer. 2010; 1:580-586. doi: 10.1177/1947601910379012.
128. Eisenman RN. Deconstructing myc. Genes Dev. 2001; 15:2023-2030.
129. Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC and Li F. The c-Myc target gene network. Seminars in cancer biology. 2006; 16:253-264.
130. Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA and Dang CV. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. The Journal of biological chemistry. 2000; 275:21797-21800.
131. Gordan JD, Thompson CB and Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer cell. 2007; 12:108-113.
132. Rimpi S and Nilsson JA. Metabolic enzymes regulated by the Myc oncogene are possible targets for chemotherapy or chemoprevention. Biochem Soc Trans. 2007; 35:305-310.
133. Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O’Donnell KA, Kim JW, Yustein JT, Lee LA and Dang CV. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Molecular and cellular biology. 2005; 25:6225-6234.
134. Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang CV and Semenza GL. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer cell. 2007; 11:407-420.
135. Morrish F, Giedt C and Hockenbery D. c-MYC apoptotic function is mediated by NRF-1 target genes. Genes Dev. 2003; 17:240-255.
136. Morrish F, Neretti N, Sedivy JM and Hockenbery DM. The oncogene c-Myc coordinates regulation of metabolic networks to enable rapid cell cycle entry. Cell Cycle. 2008; 7:1054-1066.
137. Murphy TA, Dang CV and Young JD. Isotopically nonstationary 13C flux analysis of Myc-induced metabolic reprogramming in B-cells. Metab Eng. 2013; 15:206-217.
138. Bello-Fernandez C, Packham G and Cleveland JL. The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proceedings of the National Academy of Sciences of the United States of America. 1993; 90:7804-7808.
139. Liu YC, Li F, Handler J, Huang CR, Xiang Y, Neretti N, Sedivy JM, Zeller KI and Dang CV. Global regulation of nucleotide biosynthetic genes by c-Myc. PloS one. 2008; 3:e2722.
140. Ruiz-Perez MV, Medina MA, Urdiales JL, Keinanen TA and Sanchez-Jimenez F. Polyamine metabolism is sensitive to glycolysis inhibition in human neuroblastoma cells. The Journal of biological chemistry. 2015; 290:6106-6119.
141. Celano P, Baylin SB and Casero RA, Jr. Polyamines differentially modulate the transcription of growth-associated genes in human colon carcinoma cells. The Journal of biological chemistry. 1989; 264:8922-8927.
142. Vazquez A, Markert EK and Oltvai ZN. Serine biosynthesis with one carbon catabolism and the glycine cleavage system represents a novel pathway for ATP generation. PloS one. 2011; 6:e25881.
143. Vazquez A, Tedeschi PM and Bertino JR. Overexpression of the mitochondrial folate and glycine-serine pathway: a new determinant of methotrexate selectivity in tumors. Cancer Res. 2013; 73:478-482.
144. Morrish F, Isern N, Sadilek M, Jeffrey M and Hockenbery DM. c-Myc activates multiple metabolic networks to generate substrates for cell-cycle entry. Oncogene. 2009; 28:2485-2491.
145. Bretones G, Delgado MD and Leon J. Myc and cell cycle control. Biochimica et biophysica acta. 2014.
146. Schuhmacher M, Staege MS, Pajic A, Polack A, Weidle UH, Bornkamm GW, Eick D and Kohlhuber F. Control of cell growth by c-Myc in the absence of cell division. Curr Biol. 1999; 9:1255-1258.
147. Hermeking H, Rago C, Schuhmacher M, Li Q, Barrett JF, Obaya AJ, O’Connell BC, Mateyak MK, Tam W, Kohlhuber F, Dang CV, Sedivy JM, Eick D, et al. Identification of CDK4 as a target of c-MYC. Proceedings of the National Academy of Sciences of the United States of America. 2000; 97:2229-2234.
148. Zeller KI, Jegga AG, Aronow BJ, O’Donnell KA and Dang CV. An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol. 2003; 4:R69.
149. Gregory MA and Hann SR. c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt’s lymphoma cells. Molecular and cellular biology. 2000; 20:2423-2435.
150. Sears R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes & Development. 2000; 14:2501-2514.
151. Sears R, Leone G, DeGregori J and Nevins JR. Ras enhances Myc protein stability. Mol Cell. 1999; 3:169-179.
152. Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, Counter CM, Nevins JR, Means AR, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004; 6:308-318.
153. Flinn EM, Busch CM and Wright AP. myc boxes, which are conserved in myc family proteins, are signals for protein degradation via the proteasome. Molecular and cellular biology. 1998; 18:5961-5969.
154. Sears RC. The life cycle of C-myc: from synthesis to degradation. Cell Cycle. 2004; 3:1133-1137.
155. Gordan JD and Simon MC. Hypoxia-inducible factors: central regulators of the tumor phenotype. Curr Opin Genet Dev. 2007; 17:71-77.
156. Dang CV, Kim JW, Gao P and Yustein J. The interplay between MYC and HIF in cancer. Nat Rev Cancer. 2008; 8:51-56.
157. Guzy RD and Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol. 2006; 91:807-819.
158. Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda). 2009; 24:97-106.
159. Majmundar AJ, Wong WJ and Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010; 40:294-309.
160. Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010; 29:625-634.
161. Chan DA, Sutphin PD, Denko NC and Giaccia AJ. Role of prolyl hydroxylation in oncogenically stabilized hypoxia-inducible factor-1alpha. The Journal of biological chemistry. 2002; 277:40112-40117.
162. Berra E, Benizri E, Ginouves A, Volmat V, Roux D and Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003; 22:4082-4090.
163. Hirsila M, Koivunen P, Gunzler V, Kivirikko KI and Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. The Journal of biological chemistry. 2003; 278:30772-30780.
164. Elvidge GP, Glenny L, Appelhoff RJ, Ratcliffe PJ, Ragoussis J and Gleadle JM. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: the role of HIF-1alpha, HIF-2alpha, and other pathways. The Journal of biological chemistry. 2006; 281:15215-15226.
165. MacKenzie ED, Selak MA, Tennant DA, Payne LJ, Crosby S, Frederiksen CM, Watson DG and Gottlieb E. Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Molecular and cellular biology. 2007; 27:3282-3289.
166. Tennant DA, Frezza C, MacKenzie ED, Nguyen QD, Zheng L, Selak MA, Roberts DL, Dive C, Watson DG, Aboagye EO and Gottlieb E. Reactivating HIF prolyl hydroxylases under hypoxia results in metabolic catastrophe and cell death. Oncogene. 2009; 28:4009-4021.
167. Duran RV, MacKenzie ED, Boulahbel H, Frezza C, Heiserich L, Tardito S, Bussolati O, Rocha S, Hall MN and Gottlieb E. HIF-independent role of prolyl hydroxylases in the cellular response to amino acids. Oncogene. 2013; 32:4549-4556.
168. Baek JH, Liu YV, McDonald KR, Wesley JB, Zhang H and Semenza GL. Spermidine/spermine N(1)-acetyltransferase-1 binds to hypoxia-inducible factor-1alpha (HIF-1alpha) and RACK1 and promotes ubiquitination and degradation of HIF-1alpha. The Journal of biological chemistry. 2007; 282:33358-33366.
169. Hu CJ, Wang LY, Chodosh LA, Keith B and Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Molecular and cellular biology. 2003; 23:9361-9374.
170. Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY and Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998; 12:149-162.
171. Kim JW, Tchernyshyov I, Semenza GL and Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell metabolism. 2006; 3:177-185.
172. Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV and Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007; 129:111-122.
173. Pescador N, Villar D, Cifuentes D, Garcia-Rocha M, Ortiz-Barahona A, Vazquez S, Ordonez A, Cuevas Y, Saez-Morales D, Garcia-Bermejo ML, Landazuri MO, Guinovart J and del Peso L. Hypoxia promotes glycogen accumulation through hypoxia inducible factor (HIF)-mediated induction of glycogen synthase 1. PloS one. 2010; 5:e9644.
174. Pelletier J, Bellot G, Gounon P, Lacas-Gervais S, Pouyssegur J and Mazure NM. Glycogen Synthesis is Induced in Hypoxia by the Hypoxia-Inducible Factor and Promotes Cancer Cell Survival. Front Oncol. 2012; 2:18.
175. Brahimi-Horn MC, Bellot G and Pouyssegur J. Hypoxia and energetic tumour metabolism. Curr Opin Genet Dev. 2011; 21:67-72.
176. Huang LE. Carrot and stick: HIF-alpha engages c-Myc in hypoxic adaptation. Cell Death Differ. 2008; 15:672-677.
177. Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC and Huang LE. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. Embo J. 2004; 23:1949-1956. Epub 2004 Apr 1948.
178. Koshiji M, To KK, Hammer S, Kumamoto K, Harris AL, Modrich P and Huang LE. HIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expression. Mol Cell. 2005; 17:793-803.
179. Wong WJ, Qiu B, Nakazawa MS, Qing G and Simon MC. MYC degradation under low O2 tension promotes survival by evading hypoxia-induced cell death. Molecular and cellular biology. 2013; 33:3494-3504.
180. Li Q, Kluz T, Sun H and Costa M. Mechanisms of c-myc degradation by nickel compounds and hypoxia. PloS one. 2009; 4:e8531.
181. Hubbi ME, Luo W, Baek JH and Semenza GL. MCM proteins are negative regulators of hypoxia-inducible factor 1. Molecular cell. 2011; 42:700-712.
182. Semenza GL. Hypoxia. Cross talk between oxygen sensing and the cell cycle machinery. Am J Physiol Cell Physiol. 2011; 301:C550-552.
183. Zwaans BM and Lombard DB. Interplay between sirtuins, MYC and hypoxia-inducible factor in cancer-associated metabolic reprogramming. Dis Model Mech. 2014; 7:1023-1032.
184. Zhdanov AV, Waters AH, Golubeva AV and Papkovsky DB. Differential contribution of key metabolic substrates and cellular oxygen in HIF signalling. Exp Cell Res. 2015; 330:13-28.
185. Okuyama H, Endo H, Akashika T, Kato K and Inoue M. Downregulation of c-MYC protein levels contributes to cancer cell survival under dual deficiency of oxygen and glucose. Cancer Research. 2010; 70:10213-10223.
186. Thorpe LM, Yuzugullu H and Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015; 15:7-24.
187. Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, Tsou P, Gan W, Papa A, Kim BM, Wan L, Singh A, Zhai B, et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature. 2014; 508:541-545.
188. Altomare DA and Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005; 24:7455-7464.
189. Greer EL and Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005; 24:7410-7425.
190. Wtroba M, Maliska D and Maliski S. Current overview of functions of FoxO proteins, with special regards to cellular homeostasis, cell response to stress, as well as inflammation and aging. Advances in Medical Sciences. 2012; 57.
191. Eijkelenboom A and Burgering BMT. FOXOs: signalling integrators for homeostasis maintenance. Nature Reviews Molecular Cell Biology. 2013; 14:83-97.
192. Yeo H, Lyssiotis CA, Zhang Y, Ying H, Asara JM, Cantley LC and Paik J-H. FoxO3 coordinates metabolic pathways to maintain redox balance in neural stem cells. The EMBO Journal. 2013; 32:2589-2602.
193. Shats I, Gatza ML, Liu B, Angus SP, You L and Nevins JR. FOXO transcription factors control E2F1 transcriptional specificity and apoptotic function. Cancer Res. 2013; 73:6056-6067. doi: 6010.1158/0008-5472.CAN-6013-0453. Epub 2013 Aug 6021.
194. Nho RS and Hergert P. FoxO3a and disease progression. World J Biol Chem. 2014; 5:346-354.
195. Fu Z and Tindall DJ. FOXOs, cancer and regulation of apoptosis. Oncogene. 2008; 27:2312-2319.
196. Lam EW, Brosens JJ, Gomes AR and Koo CY. Forkhead box proteins: tuning forks for transcriptional harmony. Nat Rev Cancer. 2013; 13:482-495.
197. Eijkelenboom A, Mokry M, de Wit E, Smits LM, Polderman PE, van Triest MH, van Boxtel R, Schulze A, de Laat W, Cuppen E and Burgering BM. Genome-wide analysis of FOXO3 mediated transcription regulation through RNA polymerase II profiling. Mol Syst Biol. 2013; 9:638.
198. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J and Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999; 96:857-868.
199. Biggs WH, 3rd, Meisenhelder J, Hunter T, Cavenee WK and Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proceedings of the National Academy of Sciences of the United States of America. 1999; 96:7421-7426.
200. Calnan DR and Brunet A. The FoxO code. Oncogene. 2008; 27:2276-2288.
201. Wang Y, Zhou Y and Graves DT. FOXO transcription factors: their clinical significance and regulation. Biomed Res Int. 2014; 2014:925350.
202. Gomes AR, Zhao F and Lam EW. Role and regulation of the forkhead transcription factors FOXO3a and FOXM1 in carcinogenesis and drug resistance. Chin J Cancer. 2013; 32:365-370.
203. Karadedou CT, Gomes AR, Chen J, Petkovic M, Ho KK, Zwolinska AK, Feltes A, Wong SY, Chan KY, Cheung YN, Tsang JW, Brosens JJ, Khoo US, et al. FOXO3a represses VEGF expression through FOXM1-dependent and -independent mechanisms in breast cancer. Oncogene. 2012; 31:1845-1858.
204. Yung MM, Chan DW, Liu VW, Yao KM and Ngan HY. Activation of AMPK inhibits cervical cancer cell growth through AKT/FOXO3a/FOXM1 signaling cascade. BMC Cancer. 2013; 13:327.
205. Jiang L, Cao XC, Cao JG, Liu F, Quan MF, Sheng XF and Ren KQ. Casticin induces ovarian cancer cell apoptosis by repressing FoxM1 through the activation of FOXO3a. Oncol Lett. 2013; 5:1605-1610.
206. Liu H, Yin J, Wang C, Gu Y, Deng M and He Z. FOXO3a mediates the cytotoxic effects of cisplatin in lung cancer cells. Anticancer Drugs. 2014; 25:898-907.
207. Kwok JM, Peck B, Monteiro LJ, Schwenen HD, Millour J, Coombes RC, Myatt SS and Lam EW. FOXM1 confers acquired cisplatin resistance in breast cancer cells. Mol Cancer Res. 2010; 8:24-34.
208. McGovern UB, Francis RE, Peck B, Guest SK, Wang J, Myatt SS, Krol J, Kwok JM, Polychronis A, Coombes RC and Lam EW. Gefitinib (Iressa) represses FOXM1 expression via FOXO3a in breast cancer. Mol Cancer Ther. 2009; 8:582-591.
209. Fernandez de Mattos S, Villalonga P, Clardy J and Lam EWF. FOXO3a mediates the cytotoxic effects of cisplatin in colon cancer cells. Molecular Cancer Therapeutics. 2008; 7:3237-3246.
210. Cortés R, Tarrado-Castellarnau M, Talancón D, López C, Link W, Ruiz D, Centelles JJ, Quirante J and Cascante M. A novel cyclometallated Pt(ii)-ferrocene complex induces nuclear FOXO3a localization and apoptosis and synergizes with cisplatin to inhibit lung cancer cell proliferation. Metallomics. 2014; 6:622.
211. Fang L, Wang H, Zhou L and Yu D. FOXO3a reactivation mediates the synergistic cytotoxic effects of rapamycin and cisplatin in oral squamous cell carcinoma cells. Toxicology and Applied Pharmacology. 2011; 251:8-15.
212. Dansen TB and Burgering BM. Unravelling the tumor-suppressive functions of FOXO proteins. Trends in cell biology. 2008; 18:421-429.
213. Reagan-Shaw S. RNA Interference-Mediated Depletion of Phosphoinositide 3-Kinase Activates Forkhead Box Class O Transcription Factors and Induces Cell Cycle Arrest and Apoptosis in Breast Carcinoma Cells. Cancer Research. 2006; 66:1062-1069.
214. de Keizer PL, Packer LM, Szypowska AA, Riedl-Polderman PE, van den Broek NJ, de Bruin A, Dansen TB, Marais R, Brenkman AB and Burgering BM. Activation of forkhead box O transcription factors by oncogenic BRAF promotes p21cip1-dependent senescence. Cancer Res. 2010; 70:8526-8536.
215. Zhang X, Tang N, Hadden TJ and Rishi AK. Akt, FoxO and regulation of apoptosis. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2011; 1813:1978-1986.
216. Fang L, Wang H, Zhou L and Yu D. Akt-FOXO3a signaling axis dysregulation in human oral squamous cell carcinoma and potent efficacy of FOXO3a-targeted gene therapy. Oral Oncology. 2011; 47:16-21.
217. Zanella F, dos Santos NR and Link W. Moving to the Core: Spatiotemporal Analysis of Forkhead Box O (FOXO) and Nuclear Factor-B (NF-B) Nuclear Translocation. Traffic. 2013; 14:247-258.
218. Liang J and Slingerland JM. Multiple Roles of the PI3K/PKB (Akt) Pathway in Cell Cycle Progression. Cell Cycle. 2003; 2:336-342.
219. Bouchard C, Marquardt J, Bras A, Medema RH and Eilers M. Myc-induced proliferation and transformation require Akt-mediated phosphorylation of FoxO proteins. Embo J. 2004; 23:2830-2840.
220. Ferber EC, Peck B, Delpuech O, Bell GP, East P and Schulze A. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ. 2012; 19:968-979.
221. Delpuech O, Griffiths B, East P, Essafi A, Lam EW, Burgering B, Downward J and Schulze A. Induction of Mxi1-SR alpha by FOXO3a contributes to repression of Myc-dependent gene expression. Molecular and cellular biology. 2007; 27:4917-4930.
222. Peck B, Ferber EC and Schulze A. Antagonism between FOXO and MYC Regulates Cellular Powerhouse. Front Oncol. 2013; 3:96.
223. Jensen KS, Binderup T, Jensen KT, Therkelsen I, Borup R, Nilsson E, Multhaupt H, Bouchard C, Quistorff B, Kjaer A, Landberg G and Staller P. FoxO3A promotes metabolic adaptation to hypoxia by antagonizing Myc function. Embo J. 2011; 30:4554-4570.
224. Bakker WJ, Harris IS and Mak TW. FOXO3a is activated in response to hypoxic stress and inhibits HIF1-induced apoptosis via regulation of CITED2. Mol Cell. 2007; 28:941-953.
225. Lin A, Yao J, Zhuang L, Wang D, Han J, Lam EW and Gan B. The FoxO-BNIP3 axis exerts a unique regulation of mTORC1 and cell survival under energy stress. Oncogene. 2014; 33:3183-3194.
226. Mori S, Nada S, Kimura H, Tajima S, Takahashi Y, Kitamura A, Oneyama C and Okada M. The mTOR Pathway Controls Cell Proliferation by Regulating the FoxO3a Transcription Factor via SGK1 Kinase. PloS one. 2014; 9:e88891.
227. Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T, Iwanami A, Liu F, Villa GR, Gu Y, Campos C, Zhu S, Yang H, et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell metabolism. 2013; 18:726-739.
228. Laplante M and Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012; 149:274-293.
229. Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006; 6:729-734.
230. Efeyan A and Sabatini DM. mTOR and cancer: many loops in one pathway. Curr Opin Cell Biol. 2010; 22:169-176.
231. Inoki K, Li Y, Xu T and Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003; 17:1829-1834.
232. Hay N and Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004; 18:1926-1945.
233. Laplante M and Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol. 2009; 19:R1046-1052.
234. Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK and Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007; 450:736-740.
235. Laplante M and Sabatini DM. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci. 2013; 126:1713-1719.
236. Sarbassov DD, Guertin DA, Ali SM and Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science (New York, NY. 2005; 307:1098-1101.
237. Arsham AM, Howell JJ and Simon MC. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. The Journal of biological chemistry. 2003; 278:29655-29660.
238. Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW and Kaelin WG, Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004; 18:2893-2904.
239. Wouters BG and Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008; 8:851-864.
240. Li Y, Wang Y, Kim E, Beemiller P, Wang CY, Swanson J, You M and Guan KL. Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. The Journal of biological chemistry. 2007; 282:35803-35813.
241. Ravitz MJ, Chen L, Lynch M and Schmidt EV. c-myc Repression of TSC2 contributes to control of translation initiation and Myc-induced transformation. Cancer Research. 2007; 67:11209-11217.
242. Csibi A, Lee G, Yoon SO, Tong H, Ilter D, Elia I, Fendt SM, Roberts TM and Blenis J. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr Biol. 2014; 24:2274-2280.
243. Zhao Y, Butler EB and Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013; 4:e532.
244. Riganti C, Gazzano E, Polimeni M, Aldieri E and Ghigo D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free Radical Biology and Medicine. 2012; 53:421-436.
245. Farber S and Diamond LK. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N Engl J Med. 1948; 238:787-793.
246. Heidelberger C, Chaudhuri NK, Danneberg P, Mooren D, Griesbach L, Duschinsky R, Schnitzer RJ, Pleven E and Scheiner J. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature. 1957; 179:663-666.
247. Diaz-Moralli S, Tarrado-Castellarnau M, Miranda A and Cascante M. Targeting Cell Cycle Regulation in Cancer Therapy. Pharmacol Ther. 2013; 138:255-271.
248. Yu L, Chen X, Wang L and Chen S. The sweet trap in tumors: aerobic glycolysis and potential targets for therapy. Oncotarget. 2016; 7:38908-38926. doi: 10.18632/oncotarget.7676.
249. Rais B, Comin B, Puigjaner J, Brandes JL, Creppy E, Saboureau D, Ennamany R, Lee WN, Boros LG and Cascante M. Oxythiamine and dehydroepiandrosterone induce a G1 phase cycle arrest in Ehrlich’s tumor cells through inhibition of the pentose cycle. FEBS Lett. 1999; 456:113-118.
250. Papandreou I, Goliasova T and Denko NC. Anticancer drugs that target metabolism: Is dichloroacetate the new paradigm? Int J Cancer. 2011; 128:1001-1008.
251. Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS and Petruk KC. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010; 2:31ra34.
252. Dunbar EM, Coats BS, Shroads AL, Langaee T, Lew A, Forder JR, Shuster JJ, Wagner DA and Stacpoole PW. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest New Drugs. 2014; 32:452-464.
253. Ishiguro T, Ishiguro M, Ishiguro R and Iwai S. Cotreatment with dichloroacetate and omeprazole exhibits a synergistic antiproliferative effect on malignant tumors. Oncol Lett. 2012; 3:726-728.
254. Olver IN, Green M, Millward MJ and Bishop JF. Phase II study of acivicin in patients with recurrent high grade astrocytoma. J Clin Neurosci. 1998; 5:46-48.
255. Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, Wilson KF, Ambrosio AL, Dias SM, Dang CV and Cerione RA. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer cell. 2010; 18:207-219.
256. Xiang Y, Stine ZE, Xia J, Lu Y, O’Connor RS, Altman BJ, Hsieh AL, Gouw AM, Thomas AG, Gao P, Sun L, Song L, Yan B, et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. The Journal of clinical investigation. 2015; 125:2293-2306.
257. Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS, Rabinowitz JD, Dang CV and Riggins GJ. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010; 70:8981-8987.
258. Jacque N, Ronchetti AM, Larrue C, Meunier G, Birsen R, Willems L, Saland E, Decroocq J, Thiago TT, Lambert M, Poulain L, Hospital MA, Sujobert P, et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood. 2015; 126:1346-1356.
259. Li B and Simon MC. Molecular Pathways: Targeting MYC-induced metabolic reprogramming and oncogenic stress in cancer. Clin Cancer Res. 2013; 19:5835-5841.
260. Goga A, Yang D, Tward AD, Morgan DO and Bishop JM. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat Med. 2007; 13:820-827.
261. Yang D, Liu H, Goga A, Kim S, Yuneva M and Bishop JM. Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107:13836-13841.
262. Ferreira BI, Hill R and Link W. Special Review: Caught in the Crosshairs: Targeted Drugs and Personalized Medicine. Cancer J. 2015; 21:441-447.
263. Narang A and Desai D. (2009). Anticancer Drug Development. In: Lu Y and Mahato RI, eds. Pharmaceutical Perspectives of Cancer Therapeutics: Springer US), pp. 49-92.
264. Li F, Zhao C and Wang L. Molecular-targeted agents combination therapy for cancer: developments and potentials. Int J Cancer. 2014; 134:1257-1269.
265. Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, Olson JA, Jr., Marks JR, Dressman HK, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006; 439:353-357.
266. Yoshida GJ. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res. 2015; 34:111.
267. Garraway LA and Janne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012; 2:214-226.
268. Chou TC. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Research. 2010; 70:440-446.