
INTRODUCTION
High-grade serous ovarian cancer (HGSOC) is the most lethal gynecological cancer in women and despite improved surgical techniques and drug regimens, overall survival has not changed significantly for several decades [1]. Intriguingly, there are individuals who live on for more than 10 years after diagnosis, also known as long-term survivors. However, the molecular and cellular mechanisms underlying this long-term survivorship are still unknown despite the presence of more long-term survivors than previously expected [2]. Hence, the identification of molecular pathways of long-term survival could significantly advance treatment options of HGSOC.
Almost all cancer biomarkers used in the clinics nowadays are secreted glycoproteins [3]. In particular, in ovarian cancer CA125 is the most commonly used tumor marker and is glycosylated at threonine (T) or serine (S) and asparagine (N), resulting in O- and N-glycopeptides, respectively [4]. The synthesis of N-glycosylation at the consensus sequence, Asn-X-Ser/Thr, of a polypeptide chain involves a complex biosynthetic pathway in which the core glycan Glc3Man9GlcNAc2 is trimmed by several glycosidases to form high mannose N-glycans, and further processed in the Golgi apparatus by the sequential action of GlcNAc T-I (MGAT1) to form hybrid N-glycans and subsequently by GlcNAc T-II (MGAT2) to form bi-antennary complex type N-glycans [5]. The addition of β1-4 GlcNAc to the 4-linked central mannose residue of the trimannosyl core Man3GlcNAc2 of hybrid, bi-antennary or even tri-antennary N-glycans by GlcNAc T-III (GnT-III), encoded by the MGAT3 gene, leads to the formation of bisecting GlcNAc N-glycan structures, thereby preventing further glycan branching [6]. Importantly, no other glycosyltransferase was shown to contribute to the formation of bisecting GlcNAc containing N-glycans as shown through genetic knockout studies in mice [7, 8]. In regards to ovarian cancer, the presence of this glycan motif was described in membrane and secreted glycoproteins from serous ovarian cancer specimens [9, 10] and MGAT3 expression was shown to be elevated in ovarian cancer cell lines and tissue when compared to normal counterparts [11, 12].
Mechanisms underlying MGAT3/ bisecting GlcNAc expression in cancer have not been analyzed in detail, especially in ovarian cancer. Our previous findings have indicated that the relative ion intensities of bisecting GlcNAc-type bi-antennary N-glycans correlate with MGAT3 expression in ovarian cancer cells [12]. Moreover, MGAT3 expression can be increased upon 5-aza-2′-deoxycytidine (5-Aza) treatment, indicating an epigenetic regulation of MGAT3 through DNA methylation [12]. This is in line with mouse experiments suggesting a transcriptional impact of DNA methylation at the CpG island of MGAT3 during epithelial-to-mesenchymal transition [13].
Whilst aberrant glycosylation on membrane proteins and lipids has been well documented in several cancers [14, 15], the consequence of epigenetic regulation on specific glycosylation alteration remains largely unexplored. There are a few examples in the literature on DNA methylation-based transcriptional regulation of glycosyltransferase-encoding genes with almost all of them using DNA methyltransferase inhibitor 5-Aza [12, 16–20]. However, specific examples for 5-Aza-imposed DNA methylation leading to elevated glycosyltransferase-encoding gene expression and, more importantly, to the presence of the corresponding glycan motif on glycoproteins are rather limited. One example is the epigenetic regulation of alpha 2,3 sialyltransferase 6 (ST3Gal6) and its glycosidic product Lewis x which was reconstituted by 5-Aza in the colon cancer cell line HCT15 [16] and confirmed in the serous ovarian cancer cell line OVCAR3 with no additional analysis performed on CpG island DNA methylation [19].
In this study, we investigated whether DNA methylation of MGAT3 gene in ovarian cancer can lead to the presence of corresponding N-glycosidic products bearing bisecting GlcNAc and how MGAT3 expression impacts on cancer patient survival.
RESULTS
Human MGAT3 contains two large CpG islands and is regulated by DNA methylation at the transcription starting site (TSS) in ovarian cancer cells
Our previous study suggested a correlation between bisecting GlcNAc containing N-glycoproteins and MGAT3 expression, which might be linked to the methylation status of the MGAT3 promoter in ovarian cancer cell lines [12]. However, the detailed analysis of the human MGAT3 promoter in terms of the presence of a CpG island and the DNA methylation thereof was not addressed. Moreover, the reconstitution of bisecting GlcNAc in cells with negligible MGAT3 expression upon treatment with 5-Aza was not shown. Thus, we sought to analyze the MGAT3 genomic region for the presence of CpG islands in more detail. We used four different web-based bioinformatic engines to search for a CpG island in the MGAT3 gene (UCSC Genome Browser, CpGPLOT, Methprimer and CpG island searcher [21–24]). All four programs predicted two CpG islands at the transcription start site (TSS) of the MGAT3 gene. One CpG island encompasses exon 1 and the second is located within Exon 2 of MGAT3 (Figure 1A). The length and CpG content vary depending on the program used for prediction but is within the range of 220 CpGs in 1600 bp for CpG island 1 and 120 CpGs in 1500 bp for CpG island 2 (Figure 1A, Supplementary Table 1). Since most changes in DNA methylation occur in a region of −250/+250 bp relative to the TSS [25], we analyzed the DNA methylation status around the TSS of MGAT3 (Figure 1B). The high density of CpGs in the CpG island 1 rendered it difficult to design primers devoid of CpG dinucleotides. We found one pair that amplified 230 bp (−200/+30 relative to the TSS) of which both primers did not contain CpGs (Figure 1B). The amplified region contained 38 CpGs, 31 upstream and 7 downstream of the MGAT3 TSS.
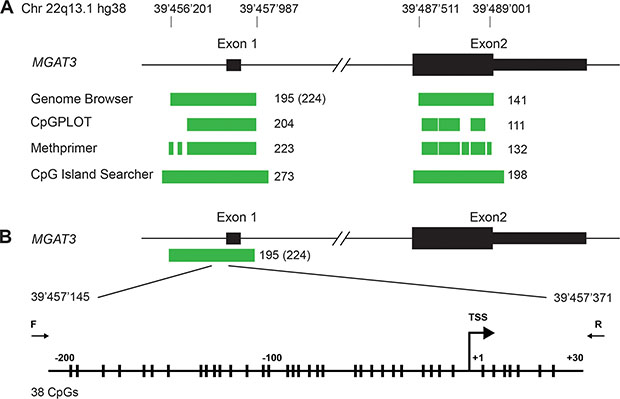
Figure 1: Human MGAT3 gene contains two CpG Islands. (A) CpG island analysis of the genomic locus of MGAT3. Exon 1 and 2 are shown in black (open reading frame depicted as larger bar), predicted CpG islands are depicted in green. (B) Genomic region analyzed in detail around the TSS is magnified. Individual CpGs are depicted with vertical bars and the TSS is indicated with a kinked arrow. Positions of forward and reverse primer used for bisulfite sequencing are indicated.
The analysis of MGAT3 promoter DNA methylation was performed in normal human ovarian surface epithelial cell lines (HOSE6-3 and HOSE17-1) and two serous ovarian cancer cell lines (OVCAR3 and A2780), showing low and high MGAT3 expression, respectively (Figure 2A). Bisulfite sequencing revealed hypermethylation of the analyzed region in HOSE6-3 and HOSE17-1 (68.1 ± 6.3% and 58.8 ± 6.8%, respectively) cells compared to hypomethylation in OVCAR3 and A2780 cells (2.7 ± 1.0% and 21.9 ± 6.1%, respectively) (Figure 2B). This demonstrates that DNA methylation around the TSS of MGAT3 correlates with gene expression.
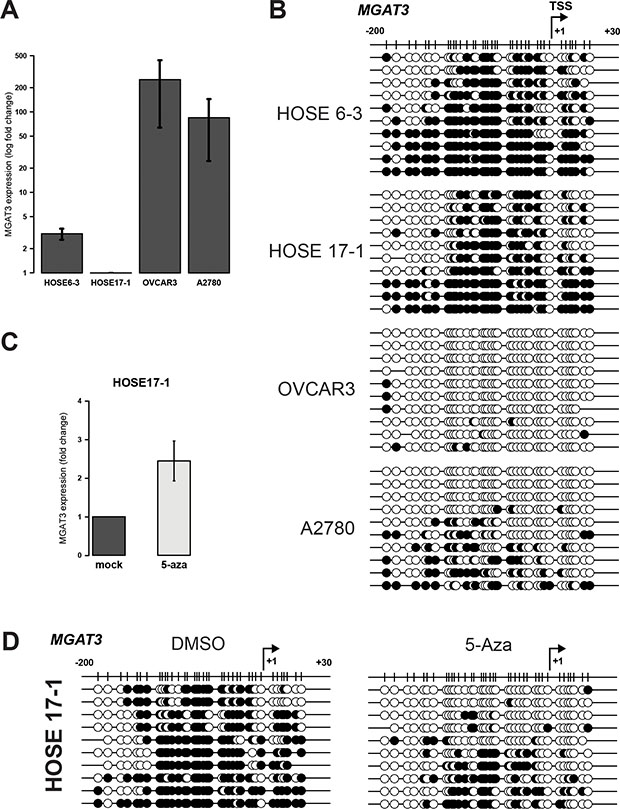
Figure 2: DNA methylation at −200/+30 from the TSS corroborates with MGAT3 expression in HOSE and ovarian cancer cells. (A) Relative MGAT3 gene expression analyzed by RT-qPCR. MGAT3 expression was normalized to logarithmic mean of three independent reference genes (HSPCB, SDHA, and YWHAZ). (B) Bisulfite sequencing result illustrated with black (methylated CpG) and white (unmethylated CpG) circles. (C) MGAT3 expression is induced by the inhibition of DNA methyltransferases (+ 5-Aza). Gene expression was analyzed by RT-qPCR 72 hours after the addition of 5-Aza. (D) Bisulfite sequencing result of the MGAT3 genomic region from HOSE17-1 cells analyzed in (C). MGAT3 expression data are provided as mean and standard deviation of three independent experiments.
We have previously shown that MGAT3 expression can be induced by inhibition of DNA methyltransferases using 5-Aza in HOSE cells [12] (Figure 2C). Importantly, here we demonstrate that overall DNA methylation around the TSS was reduced upon treatment with 5-Aza in HOSE17-1 cells (mock 67.8 ± 3.9% versus 5-Aza 26.6 ± 6.1%, p = 0.0162), indicating a direct relationship between TSS methylation and MGAT3 expression (Figure 2D).
DNA methylation at the TSS regulates expression of MGAT3 and bisecting GlcNAc in ovarian cancer cell lines
Next, we addressed the question whether the regulation of MGAT3 expression by DNA methylation of the TSS is a common feature of ovarian cancer cells. We analyzed MGAT3 expression and the corresponding DNA methylation in three additional serous ovarian cancer cell lines. High MGAT3 expression was observed in OVCAR4 and BG-1 cells, whereas OVCAR8 cells showed unexpectedly low levels of MGAT3 expression (Figure 3A).
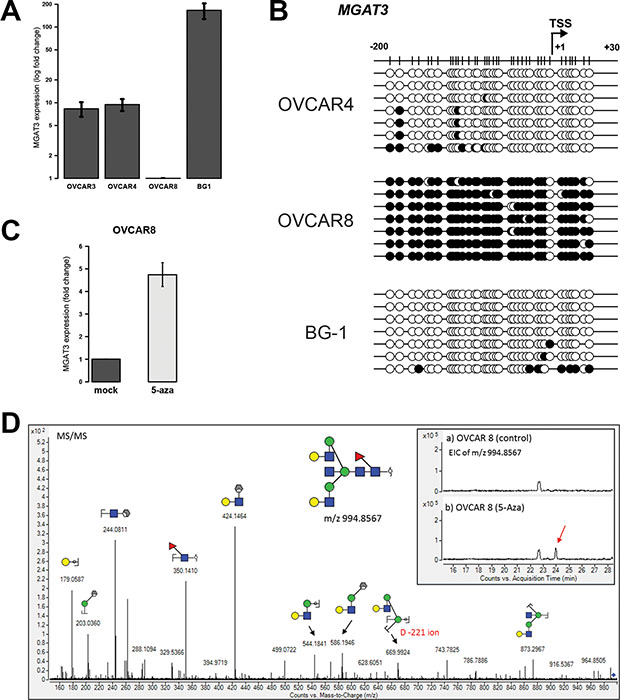
Figure 3: DNA methylation at -200/+30 from the TSS correlates with MGAT3 expression in other ovarian cancer cells and bisecting GlcNAc is induced by DNA methyltransferase inhibition. (A) MGAT3 gene expression analysis by RT-qPCR. (B) Bisulfite sequencing result of corresponding cell lines profiled in A. Black circle (methylated CpG), white circle (unmethylated CpG). (C) MGAT3 gene expression in OVCAR8 cells was analyzed by RT-qPCR after 72 hours of treatment with 5-Aza. (D) Reconstitution of bisecting GlcNAc upon 5-Aza treatments in OVCAR8 cells. N-Glycans were PNGase F-released from OVCAR8 membrane proteins used in (C) and analyzed by ESI-qTOF mass spectrometry. MGAT3 expression data are provided as mean and standard deviation of three independent experiments.
In line with MGAT3 expression data, TSS DNA hypomethylation was observed in cell lines with high MGAT3 expression, BG-1 (3.8 ± 3.2%) and OVCAR4 (5.3 ± 2.4%) whereas DNA hypermethylation corroborate with decreased MGAT3 expression in OVCAR8 (93.2 ± 1.0%) cells (Figure 3B). Inhibition of DNA methyltransferases by 5-Aza treatment resulted in an increased expression of MGAT3 in OVCAR8 cells (Figure 3C). Importantly, mass spectrometric analysis of N-glycans released from membrane glycoproteins revealed the presence of a bisecting GlcNAc-containing glycan structure with m/z 994.8567, which was induced in OVCAR8 cells by the addition of 5-Aza (Figure 3D, insert). The presence of the bisecting GlcNAc was further verified using MS/MS fragmentation whereby the D-221 fragment ion at m/z 669.99, previously reported to be diagnostic for bisecting biantennary GlcNAc-type N-glycans [12], was found to be present in the MS/MS spectra (Figure 3D). Bisecting GlcNAc-containing glycan structures which were shown to be present on other ovarian cancer cells [26] were not detected (Supplementary Figure 2).
We next expanded our analysis to investigate whether this strong association of DNA methylation, MGAT3 gene and bisecting GlcNAc expression can be translated also to non-ovarian cancer cell lines of which N-glycan profiles are present. Therefore, we analyzed MGAT3 expression in the cervical cancer cell line, HeLa and the leukemia cell line, K562, showing differential MGAT3 expression (Figure 4A). In full concordance with gene expression, the TSS of MGAT3 was highly methylated in HeLa (93.7 ± 0.9%) and hypomethylated in K562 (3.2 ± 0.8%) cells (Figure 4B). Interestingly, this pattern fully corroborates with the absence (HeLa) or presence (K562) of bisecting GlcNAc containing N-glycans as shown by glycoprofiling data obtained through the Consortium for Functional Glycomics (CFG, www.functionalglycomics.org) (Figure 4C), hence indicating a direct link between DNA methylation and bisecting GlcNAc expression, as shown in these two non-ovarian cancer cell lines.
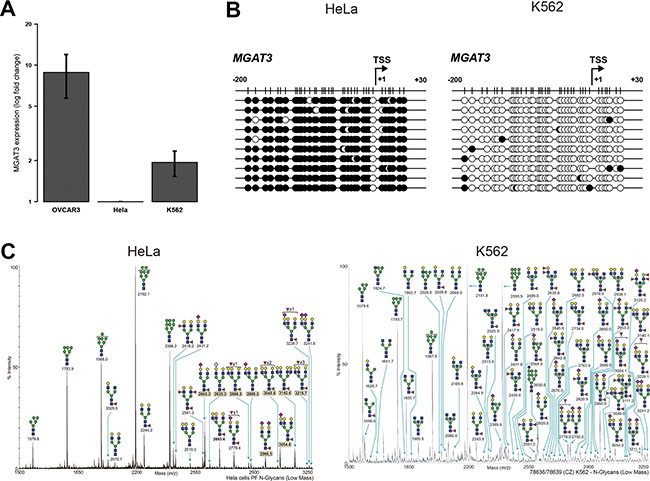
Figure 4: DNA methylation at -200/+30 from the TSS correlates with MGAT3 and bisecting GlcNAc expression in non-ovarian cancer cell lines. (A) Gene expression analysis by RT-qPCR. (B) Bisulfite sequencing showing black circle (methylated CpG) and white circle (unmethylated CpG). (C) The annotated N-glycomes of HeLa and K562 cells were accessed and downloaded through the Consortium for Functional Glycomics (CFG, www.functionalglycomics.org). Low molecular weight N-glycans are depicted.
Ovarian cancer long-term survivors show lowest MGAT3 expression
Based on the strong relationship observed between DNA methylation, MGAT3 gene and bisecting GlcNAc expression in various normal and cancer cell lines, we sought to reproduce our results in human cancer tissue samples. We analyzed the overall MGAT3 expression among all cancers ≤ (n = 12) in the TCGA PANCAN12 dataset to address the question whether MGAT3 expression is cancer-type specific (Figure 5). The MGAT3 expression varied among all TCGA datasets from being the lowest in head & neck squamous cell carcinoma to being the highest in HGSOC. Moreover, the TCGA HGSOC cohort showed significantly elevated MGAT3 expression compared to all remaining cancer types except for glioblastoma (p < 0.001, Figure 5A).
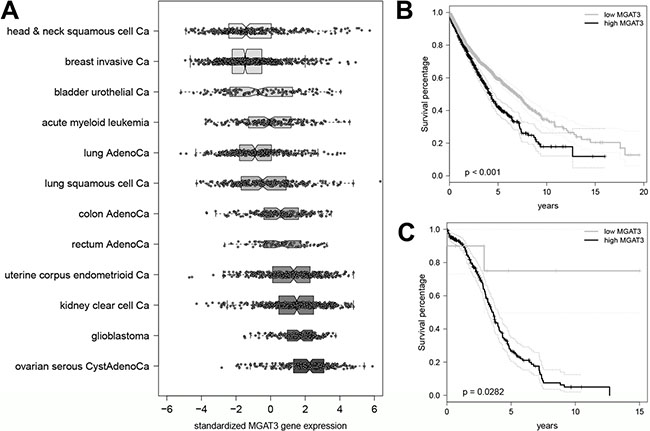
Figure 5: Highest expression of MGAT3 in HGSOC among twelve TCGA datasets. (A) Boxplot of MGAT3 expression in TCGA PANCAN12 data set (n = 3587) sorted by median gene expression from lowest (head and neck squamous cell cancer) to highest (ovarian serious cystadenocarcinoma). Boxes for each data set are drawn with widths proportional to the square roots of the number of observations in the groups. (B) Elevated MGAT3 expression is associated with poor survival in all cancer types. Kaplan-Meier curve for dichotomized MGAT3 expression in TCGA PANCAN12 data set (n = 3477). (C) Kaplan-Meier curve for dichotomized MGAT3 expression in HGSOC patients obtained from the TCGA PANCAN12 data set (n = 264).
Since ovarian cancer is the gynecological cancer with the poorest prognosis and displayed highest MGAT3 expression, we investigated whether MGAT3 expression correlates with survival in all TCGA PANCAN12 cancer samples and in particular, HGSOC (Figure 5B and 5C). The tree-based model selected a threshold (MGAT3 ≤ 1.67) in regards to survival separating all cancers (n = 3477) in low and high MGAT3 expression (p < 0.001) (Figure 5B). Interestingly, the investigation of the HGSOC cohort (n = 264) revealed a small number of patients (n = 10) which showed lowest MGAT3 expression (MGAT3 ≤ –1.177) and displayed a flattened out survival curve, indicating long-term survivorship of up to 15 years (p = 0.0282, Figure 5C). Multivariate analysis identified MGAT3 expression as an independent prognostic marker (p = 0.047, HR = 1.118, 95% CI 1.000–1.248) for overall survival. Residual disease and age at diagnosis but not FIGO stage, grade, lymphatic as well as venous invasion were identified as independent prognostic markers.
Basal-like breast cancers also display high MGAT3 expression with inverse correlation to DNA methylation
We next analyzed the correlation between DNA methylation and MGAT3 expression in human tumors in more detail. The 450K DNA methylation data (Infinium HumanMethylation450 BeadChip, Illumina, San Diego, United State of America) is not yet available for HGSOC and the 27K human DNA methylation data provided only one probe (cg18399321) for HGSOC, which is completely de-methylated (Supplementary Figure 1). Therefore we unfortunately had to exclude HGSOC from this analysis. Since recent genomic and transcriptomic data of basal-like breast cancers suggest a close molecular relation to HGSOC [27], we further analyzed MGAT3 expression in the TCGA breast cancer dataset (n = 514) and compared basal-like (n = 98), HER2-enriched (n = 58), luminal A (n = 231) and B (n = 127) types of breast cancers. Interestingly, we observed significantly elevated MGAT3 expression (ANOVA p < 0.001) in basal-like cancers compared to all other breast cancer subtypes and similar MGAT3 expression levels as in HGSOC (Figure 6A, Supplementary Table 2).
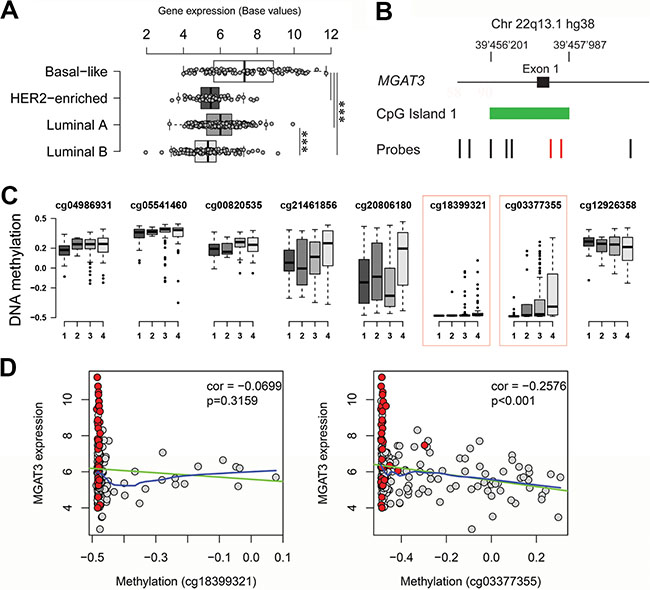
Figure 6: Elevated MGAT3 expression correlates with DNA hypomethylation in TCGA breast cancer data. (A) Boxplot showing significantly elevated MGAT3 expression in basal-like breast cancer compared to HER2-enriched, luminal A and luminal B breast cancer (***p-value < 0.001). (B) Genomic localization of probes of the 450K HumanMethylation Bead chip (black and red vertical bars) at exon 1 (black box) of MGAT3. CpG island 1 is shown in green. (C) Boxplot displaying degree of DNA methylation (normalized by centering to 0) in the TCGA breast cancer cohort for selected probes (same order left to right as annotated in A). Degree of DNA methylation is shown for basal-like (1), HER2-enriched (2), luminal-A (3), and luminal-B (4) breast cancer (abscissa). Probes significantly discriminating four breast cancer groups are highlighted in the red dotted box. (D) Correlation plot for TCGA breast cancer samples matching MGAT3 expression and DNA methylation of probes cg18399321 and cg03377355 (red highlighted in A and B). Basal-like are highlighted in red while remaining breast cancer samples are grey. Correlation and p-value are shown in each plot. Linear and locally weighted polynomial regressions are shown in green and blue, respectively.
The breast cancer dataset was also analyzed by HumanMethylation450 BeadChip data in regards to DNA methylation and MGAT3 expression in matched tissue samples. The location of the CpG probes in the 450K DNA methylation dataset was dispersed over CpG island 1 of MGAT3 (Figure 6B). We then analyzed the methylation scores of the indicated individual probes in breast cancer separated by the four subtypes. The red boxed probes (cg18399321 and cg03377355), which are in close proximity to the genomic region analyzed by bisulfite sequencing in cell lines above, displayed changes in methylation score when we compared basal-like (Lane 1) with HER2 enriched (lane 2), Luminal A (lane 3) and Luminal B (lane 4) (Figure 6C). A significant reduction of DNA methylation was observed in basal-like samples when compared to the remaining three breast cancer subtypes for probes cg18399321 (ANOVA p = 0.0189) and cg03377355 (ANOVA p < 0.001). Surrounding probes presented equally high degrees of DNA methylation among all four breast cancer subtypes (Figure 6C, Supplementary Table 2).
We then addressed the correlation between probe methylation score and MGAT3 expression in four breast cancer types. Basal-like samples were almost completely hypomethylated. A negative correlation was found for both probes with only cg03377355 being statistically significant (p < 0.001), indicating that the genomic region at this site of MGAT3 may also be involved in epigenetic regulation of MGAT3 expression (Figure 6D, Supplementary Table 2).
MGAT3 expression inversely correlates with DNA methylation in human cancer samples
Since we observed a correlation between MGAT3 expression and DNA methylation in breast cancer datasets, we investigated whether this finding can be translated to other human cancers (Figure 7). Genomic data from 18 TCGA epithelial cancer datasets were analyzed where both DNA methylation and matched MGAT3 expression data were available (n = 6118 samples). The kidney chromophobe cohort had the lowest sample size (n = 66), while the invasive breast carcinoma displayed the cohort with the largest number of samples (n = 735) in this study. In concordance with our results on cell lines as well as TCGA breast cancer dataset analysis, samples with DNA hypermethylation had generally lower levels of MGAT3 expression in the TCGA datasets. In contrast, DNA hypomethylated samples revealed a trend towards elevated MGAT3 expression (Figure 7), thus resulting in an inverse correlation for MGAT3 expression (multiple R2 0.2945; p = 0.01997).
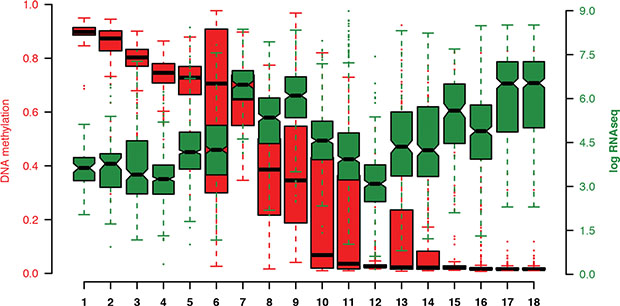
Figure 7: Boxplots of methylation values sorted by median (red) and MGAT3 expression (green) obtained by 450K DNA methylation array and RNA sequencing through the cBioPortal for cancer genomics, respectively. Each boxplot represents values obtained from the TCGA database of matched DNA methylation (red) and RNAseq data (green). DNA methylation data represent as the beta-value of the probe that is most negatively correlated with the expression of the MGAT3 gene. Black horizontal lines represent the median with the box representing the 25th and 85th percentiles, the whiskers the 5th and 95th percentiles, outliers shown by dots in corresponding color (red, green). 1) Kidney chromophobe, 2) Adrenocortical carcinoma, 3) Kidney renal papillary carcinoma, 4) Prostate carcinoma, 5) Breast invasive carcinoma, 6) Skin cutaneous melanoma, 7) Pancreatic adenocarcinoma, 8) Colorectal adenocarcinoma, 9) Kidney renal clear cell carcinoma, 10) Lung adenocarcinoma, 11) Liver hepatocellular carcinoma, 12) Cervical squamous cell carcinoma and endocervical adenocarcinoma, 14) Bladder urothelial carcinoma, 15) Uterine corpus endometrial carcinoma, 16) Lung squamous cell carcinoma, 17) Papillary Thyroid carcinoma, 18) Thyroid carcinoma.
DISCUSSION
Alterations in protein glycosylation occur frequently in cancer and can regulate tumor development and progression. Aberrant glycans might be explained by changes of glycosyltransferase gene expression, chaperone function, mislocalization of glycosyltransferases, un-/availability of sugar donors and altered glycosidase activity [28, 29]. Our current data supports the relationship of epigenetic regulation on MGAT3 expression through DNA methylation and the presence of bisecting GlcNAc on N-glycans in HGSOC. We expanded our study to epithelial cancer types other than HGSOC, demonstrating that DNA methylation and MGAT3 expression corroborates among almost all epithelial cancer types and has influence on disease outcome, independent of the cancer subtype. This relationship can be reproduced in basal-like breast cancer, a histotype resembling the molecular signature of HGSOC.
To our knowledge, this is the first detailed analysis of human MGAT3 gene which illustrates two CpG islands located in both exons consisting of roughly 200 and 100 individual CpGs, sharing similarities with the organization of the mouse MGAT3, despite being smaller in size than the human counterpart [13]. In contrast, human MGAT5 does not have a CpG island [19], whereas mouse MGAT5 has one CpG island at exon 1 [13], indicating that MGAT3 might have an evolutionary conserved type of epigenetic regulatory mechanism.
We have previously shown that the product of GnT-III (encoded by the MGAT3 gene), bisecting GlcNAc, is present on ovarian cancer cells but is absent on human ovarian surface epithelial cells [12]. We thus hypothesized that the differential gene and glycan expression might be regulated by DNA methylation. Since epigenetic changes primarily occur in close proximity to the TSS [25], we specifically analyzed DNA methylation around the TSS of MGAT3. Elevated DNA methylation correlated with decreased MGAT3 expression in human ovarian surface epithelial and vice versa in ovarian cancer cells. Moreover, the MGAT3 expression in hypermethylated cells was reconstituted upon treatment with 5-Aza, correlating with loss of DNA methylation at the TSS. We also confirmed our findings using a broader panel of ovarian and non-ovarian cancer cells. Importantly, addition of 5-Aza not only restored MGAT3 expression in OVCAR8 cells, but also resulted in the expression of bisecting GlcNAc containing N-glycans, thereby clearly indicating a direct relationship of DNA methylation-dependent bisecting GlcNAc expression. Interestingly, we did not detect a diverse range of bisecting GlcNAc-containing N-glycan structures in OVCAR8, apart from the non-sialylated bi-antennary bisecting GlcNAc N-glycan, which was also reported to be the most prominent form analyzed previously in other ovarian cancer cell lines such as OVCAR3 [26]. This demonstrates that the regulation of MGAT3 seems to be unique amongst glyco-genes involved in the N-glycosylation pathway, since other genes (MGAT4A and MGAT5) were shown to be indirectly regulated by DNA methylation [30]. A similar regulatory mechanism was only observed in the case of B4GALNT2, an enzyme involved in the synthesis of blood group carbohydrate antigens [17]. B4GALNT2 promoter DNA hypermethylation in colorectal cancer correlated with decreased gene expression and the absence of the glycan Sda on the cell surface. Similar findings were also observed for ST3GAL6 [16]. However, the promoter of B4GALNT2 and ST3GAL6 seems to be hypermethylated in cancer in contrast to the MGAT3 promoter, which is hypomethylated.
Recent genomic and transcriptomic data suggest that basal-like breast and HGSOC are intriguingly similar in terms of TP53, BRCA1/2 and RB1 mutation status and also based on transcriptional correlations [27]. In addition to that, clinicopathological characteristics are in full concordance with this molecular phenotype and both cancer types are highly aggressive [31]. Interestingly, an inverse correlation between DNA methylation of MGAT3 and gene expression was also observed in basal-like breast cancer. Since this data indicates a tissue-specific regulation of MGAT3/bisecting GlcNAc expression in human cancers, the role of MGAT3/bisecting GlcNAc in cancer development or metastasis is very likely to differ within different types of human cancers. Although bisecting GlcNAc was shown to be involved in E-cadherin- [32], epidermal growth factor (EGF)- [33], Wnt/ beta-catenin- [34] and integrin/focal adhesion kinase (FAK)- [35] mediated signaling pathways. However, underlying molecular mechanisms altered by MGAT3/bisecting GlcNAc in HGSOC are yet to be identified, for instance with the help of CRISPR/ Cas9-mediated deletion of MGAT3 in HGSOC cells.
Overall, our data demonstrate, for the first time, a direct relationship between DNA methylation around the TSS of MGAT3, MGAT3 gene expression and the presence of bisecting GlcNAc in HGSOC and basal-like breast cancer. It is evident that epigenetic regulation through DNA methylation may primarily be associated with MGAT3/ bisecting GlcNAc expression, however, other mechanisms such as nucleosome positioning [36] and histone modifications [37] are also possible.
The prognosis of HGSOC has not changed over the past decades despite advances in aggressive cytoreduction, targeted therapy and increasing knowledge about underlying genetic mutations. In fact, it still remains as the gynecological cancer with the worst prognosis, reflected by a five-year survival of only 20%. However, there are instances where HGSOC patients are present with a long-term survivorship of over ten years. To date, only few analyses, including our own, have examined this patient cohort despite its vast importance. Our analysis in regards to MGAT3 expression of patients from the large TCGA dataset indicates that the loss of MGAT3 results in a dramatic plateau in the survival curves of HGSOC. This observation has, so far, not been associated with any biomarker or treatment measure in ovarian cancer. Therefore, our data strongly supports further investigation into the role of MGAT3 as a modulator of long-term survival, reflecting a yet unknown underlying biology.
MATERIALS AND METHODS
CpG island analysis
Sequence information of human MGAT3 (Hg38, Chr22: 39448663-39498392) was extracted from the UCSC genome browser and subjected to Genome Browser, CpGPLOT, Methprimer and CpG island searcher [21–24] in order to predict CpG islands in this region using default settings.
Cell culture and treatments
HOSE6-3, HOSE17-1, OVCAR3, A2780, OVCAR4, OVCAR8, BG-1, HeLa and K562 cells were cultured in RPMI supplemented with 10% fetal bovine serum and penicillin/streptomycin. Cells were treated with 5-aza-2′-deoxycytidine (5-Aza) as described previously [38]. All cell line-based experiments were performed independently on three different passages. Cells were tested routinely for mycoplasma infection.
Reverse-transcription quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from exponentially growing cells using the ReliaPrep™ RNA Cell Miniprep system according to the manufacturers’ instructions (Promega, Dübendorf, Switzerland). Total RNA (1μg) was reverse-transcribed using iScript cDNA synthesis kit (Bio-Rad Laboratories AG, Cressier, Switzerland). RT-qPCR was performed on MGAT3 and reference genes HSPCB, SDHA, and YWHAZ in 10 μl reactions containing 10 ng cDNA (initial total RNA), 400nM forward and reverse primer, nuclease free water and 1× SensiFast™ with low ROX as the reference dye (Bioline, Biolabo scientific instruments, Châtel-St-Denis, Switzerland) on a ViiA™ 7 Real-Time PCR System (Applied Biosystems, Thermo Fisher Scientific, Reinach, Switzerland). Quantitative PCR was performed in triplicates and analyzed as recently described [26, 39].
Bisulfite sequencing
Genomic DNA was extracted from exponentially growing cells as described [38]. An amount of 1 μg genomic DNA was bisulfite converted using the EZ Methylation-Gold Kit (Zymo Research Inc., Lucerna-Chem AG, Luzern, Switzerland). Bisulfite converted DNA was subjected to PCR reaction using MyTaq™ HS DNA polymerase (Bioline, Biolabo scientific instruments, Châtel-St-Denis, Switzerland). Primers devoid of CG dinucleotides for bisulfite sequencing were designed using bisulfite primer seeker (http://www.zymoresearch.com/tools/bisulfite-primer-seeker). PCR reactions contained: 1× MyTaq reaction buffer, MyTaq HS polymerase, 300 nM forward primer (5′-gggggaggggagggggtt-3′), 300 nM reverse primer (5′-cctccccccacccccacttc-3′) and nuclease free water to a final volume of 10 μl. PCR was performed by initial denaturation at 95°C for 1 min, followed by 40 cycles of 95°C for 15 sec, 66°C for 15 sec, 72°C for 20 sec and a final amplicon elongation at 72°C for 10 min. The PCR product (230 bp) was visualized on a 2% agarose gel and purified from the gel using the Wizard SV Gel and PCR clean up system (Promega, Dübendorf, Switzerland). The purified PCR product was cloned into pGEM®-T Easy (Promega) and plasmid minipreps (PureYield™ Plasmid MiniPrep System, Promega) from individually grown colonies were sequenced using either T7 or SP6 primer (Sanger DNA sequencing service, Source Bioscience, Berlin, Germany). Bisulfite sequencing data were analyzed and visualized using the QUMA web-based software tool [40]. Results are provided as mean ± standard error of methylated CpGs between DNA sequences. Student t-test was applied for statistical analysis.
Membrane protein extraction and release of N-glycans from OVCAR8 cells
Cell membranes of OVCAR8 cells were prepared as previously described [12]. Briefly, approximately 1 × 106 OVCAR 8 and 5-Aza-treated OVCAR8 cells were washed with PBS, pelleted through centrifugation and re-suspended with lysis buffer (50 mM Tris-HCl, 100 mM NaCl, 1 mM EDTA and protease inhibitor) at pH 7.4 prior to homogenization. Cellular debris and unlysed cells were removed by centrifugation and the supernatant was subjected to ultracentrifugation at 120,000 g to pellet the cell membranes. Cell membranes were re-suspended in Tris binding buffer containing Triton ×-114 for phase partitioning of membrane proteins. The detergent layer containing the membrane proteins were acetone-precipitated and solubilized in 8 M urea. N-glycans were prepared and purified as previously described [12]. Briefly, membrane proteins and glycoprotein standard (fetuin) were spotted onto a polyvinylidene diflouride (PVDF) membrane, air-dried overnight at room temperature and stained. Protein spots were placed in a 96-well microtiter plate and treated with PNGase F enzyme to release the N-glycans. Released glycans were collected and treated with 100 mM ammonium acetate (pH 5.0) to ensure a complete regeneration of the reducing terminus. The released N-glycans were reduced to alditols with 2M NaBH4 in 50 mM KOH and the reduction was quenched using glacial acetic acid. N-glycan alditols were purified using cation exchange chromatography and subsequent washings with methanol were performed for the removal of residual borate. The purified N-glycan alditols were re-suspended in 15 μl of MilliQ water prior to mass spectrometry analysis.
LC-ESI-qTOF-MS/MS of released N-glycan alditols
The analysis of released N-glycan’s was performed using an Agilent UHPLC system, consisting of a binary pump, autosampler, thermostat and column oven (Agilent Series 1290, Agilent Technologies, Waldbronn, Germany) connected to a high-resolution hybrid mass spectrometer (MS) (Agilent Series 6540 Q-TOF, Agilent Technologies, Santa Clara, CA, USA). The front end of the MS was equipped with a jet stream electrospray ionization source (ESI) (Agilent Technologies, Santa Clara, CA, USA). The chromatographic separation of N-glycans was modified from Anugraham et al., 2014 and applied to a conventional HPLC method. In brief, 15 μL of samples were injected onto a Hypercarb porous graphitized carbon capillary column (3 μm particle size; 2.1 × 150 mm, Thermo Fisher Scientific, Runcorn, UK). The mobile phase was made up of 10 mM ammonium bicarbonate (A) and 9:1 acetonitrile/water with 10 mM ammonium bicarbonate (B). The column was re-equilibrated with starting mobile phase condition for 8 minutes and the column compartment was kept at 40°C. The separation of N-glycans was carried out over a linear gradient method (0–5 min; 1 % B, 40 min; 25 % B; 42 min; 50 % B and 42.5 min; 80 % B, hold for 1.5 min) with a column flow of 0.4 mL/min. The ESI source was operated in negative mode with the following parameter settings: nebulizer pressure-35 psig, nozzle voltage- 0 V, sheath gas flow- 11 L/min, sheath gas temperature -375°C, drying gas flow- 8 L/min, drying gas temperature- 250°C, capillary voltage and fragmentor voltage at 4000 V and 175 V, respectively. To obtain the highest mass accuracy, a constant flow of internal reference masses (m/z 119.0363 and 981.9956) was applied on the end plate of the ESI Source. MS scan (m/z 100-1700 at 1.5 Hz) and MS/MS (m/z 100-1700 at 3 Hz) spectra were acquired with triggered data-dependent algorithm with static exclusion range of m/z 100-600 in 2 GHz mode. Precursor ions were isolated with a width of m/z 1.3 in Q1 and fragmented with a variable collision energy based on slop x (m/z) /100 + offset (slop = 3, offset = 2). The precursor ion threshold was set to 10 000 counts with a maximum of 15 precursor ions per cycle (cycle time 5.8 sec).
The MS data were analyzed with MassHunter B.06.00 (Agilent) and the monosaccharide compositions of monoisotopic masses were determined using GlycoMod tool (http://web.expasy.org/glycomod/) with a mass error of ± 20 ppm [41]. The proposed bisecting GlcNAc N-glycan structural feature was assigned and interpreted based on diagnostic MS/MS fragmentation patterns previously reported for negative ion mode MS [12] and annotated using GlycoWorkBench 2.1 software [42] . In addition, blank samples and fetuin samples which served as quality control for sample preparation were randomly measured within each sequence run and checked for background contamination.
TCGA data analysis
TCGA data sets were accessed through the UCSC Cancer Genomics Browser website [43, 44] (https://genome-cancer.ucsc.edu/proj/site/hgHeatmap/). Gene expression and matched DNA methylation (Figure 7) indicating the RNA sequencing data and Beta-value [45] respectively, were accessed using the cBioPortal for Cancer Genomics [46] using cgdsr, a R-based application programming interface (API) that provides a basic set of R functions for querying the Cancer Genomics Data Server (CDGS) hosted by the Memorial-Sloan-Kettering Cancer Center (MSKCC).
MGAT3 gene expression was analyzed using TCGA_PANCAN12_exp_HiSeqV2 dataset. MGAT3 gene expression and methylation were analyzed using the breast cancer dataset TCGA_BRCA_exp_HiSeqV2 and TCGA_BRCA_hMethyl450 (Infinium HumanMethylation 450K).
Overall survival dependent on MGAT3 expression was investigated using Kaplan-Meier curves. MGAT3 expression was dichotomized using tree based model partitioning (“ctree” in R package “party”). Computations of the predicted survivor curves were used for Cox proportional hazards regression model (R package “survival”). Multivariate analysis was performed with fitted Cox proportional hazards regression model (R package “survival”) including MGAT3 expression, FIGO stage, grade, residual disease, lymphatic invasion, venous invasion, and age at initial diagnosis. MGAT3 expression was adjusted for these covariates. Results were presented as hazard ratios (HR) with 95% confidence interval (CI) and corresponding p-values. In order to explore associations between DNA methylation and MGAT3 expression, Pearson correlation was calculated.
Comparisons between cancer subgroups were performed using one-way ANOVA. A p value < 0.05 was considered significant. All statistical evaluations were done using the statistical software R version 3.1.3 (www.R-project.org).
ACKNOWLEDGMENTS AND FUNDING
This work used publicly available glycan profiling data provided by the Consortium for Functional Glycomics (CFG). We acknowledge the cBioPortal for Cancer Genomics (http://cbioportal.org) and the TCGA research Network (http://cancergenome.nih.gov) for providing datasets.
CONFLICTS OF INTEREST
All authors declare that they have no conflicts of interest.
FUNDING
This work was supported by Swiss National Foundation (310030_143619 and 32) to V.H.S., Oncosuisse Grant (KFS_3013-08-2012) to V.H.S., and Krebsliga beider Basel 06-2013/14-2015 to V.H.S and F.J., respectively.
REFERENCES
1. Bowtell DD, Böhm S, Ahmed AA, Aspuria P-J, Bast RC, Beral V, Berek JS, Birrer MJ, Blagden S, Bookman MA, Brenton JD, Chiappinelli KB, Martins FC, et al. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer. 2015; 15:668–679.
2. Cress RD, Chen YS, Morris CR, Petersen M, Leiserowitz GS. Characteristics of Long-Term Survivors of Epithelial Ovarian Cancer. Obstet Gynecol. 2015; 126:491–497.
3. Christiansen MN, Chik J, Lee L, Anugraham M, Abrahams JL, Packer NH. Cell surface protein glycosylation in cancer. Proteomics. 2014; 14:525–546.
4. Marcos-Silva L, Narimatsu Y, Halim A, Campos D, Yang Z, Tarp MA, Pereira PJB, Mandel U, Bennett EP, Vakhrushev SY, Levery SB, David L, Clausen H. Characterization of binding epitopes of CA125 monoclonal antibodies. J Proteome Res. 2014; 13:3349–3359.
5. Taniguchi N, Korekane H. Branched N-glycans and their implications for cell adhesion, signaling and clinical applications for cancer biomarkers and in therapeutics. BMB Rep. 2011; 44:772–781.
6. Miwa HE, Song Y, Alvarez R, Cummings RD, Stanley P. The bisecting GlcNAc in cell growth control and tumor progression. Glycoconj J. 2012; 29:609–618.
7. Bhattacharyya R, Bhaumik M, Raju TS, Stanley P. Truncated, inactive N-acetylglucosaminyltransferase III (GlcNAc-TIII) induces neurological and other traits absent in mice that lack GlcNAc-TIII. The Journal of biological chemistry. 2002; 277:26300–26309.
8. Bhaumik M, Seldin MF, Stanley P. Cloning and chromosomal mapping of the mouse Mgat3 gene encoding N-acetylglucosaminyltransferase III. Gene. 1995; 164:295–300.
9. Allam H, Aoki K, Benigno BB, McDonald JF, Mackintosh SG, Tiemeyer M, Abbott KL. Glycomic analysis of membrane glycoproteins with bisecting glycosylation from ovarian cancer tissues reveals novel structures and functions. J Proteome Res. 2015; 14:434–446.
10. Zhang X, Wang Y, Qian Y, Wu X, Zhang Z, Liu X, Zhao R, Zhou L, Ruan Y, Xu J, Liu H, Ren S, Xu C, et al. Discovery of specific metastasis-related N-glycan alterations in epithelial ovarian cancer based on quantitative glycomics. PLoS One. 2014; 9:e87978.
11. Abbott KL, Nairn AV, Hall EM, Horton MB, McDonald JF, Moremen KW, Dinulescu DM, Pierce M. Focused glycomic analysis of the N-linked glycan biosynthetic pathway in ovarian cancer. Proteomics. 2008; 8:3210–3220.
12. Anugraham M, Jacob F, Nixdorf S, Everest-Dass AV, Heinzelmann-Schwarz V, Packer NH. Specific glycosylation of membrane proteins in epithelial ovarian cancer cell lines: glycan structures reflect gene expression and DNA methylation status. Mol Cell Proteomics. 2014; 13:2213–2232.
13. Pinho SS, Oliveira P, Cabral J, Carvalho S, Huntsman D, Gärtner F, Seruca R, Reis CA, Oliveira C. Loss and recovery of Mgat3 and GnT-III Mediated E-cadherin N-glycosylation is a mechanism involved in epithelial-mesenchymal-epithelial transitions. PLoS One. 2012; 7:e33191.
14. Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nature reviews Cancer. 2015; 15:540–555.
15. Hakomori S. Glycosylation defining cancer malignancy: new wine in an old bottle. Proceedings of the National Academy of Sciences of the United States of America. 2002; 99:10231–10233.
16. Chachadi VB, Cheng H, Klinkebiel D, Christman JK, Cheng P-W. 5-Aza-2′-deoxycytidine increases sialyl Lewis X on MUC1 by stimulating β-galactoside:α2,3-sialyltransferase 6 gene. Int J Biochem Cell Biol. 2011; 43:586–593.
17. Kawamura YI, Toyota M, Kawashima R, Hagiwara T, Suzuki H, Imai K, Shinomura Y, Tokino T, Kannagi R, Dohi T. DNA hypermethylation contributes to incomplete synthesis of carbohydrate determinants in gastrointestinal cancer. Gastroenterology. 2008; 135:142–151.e143.
18. Poeta ML, Massi E, Parrella P, Pellegrini P, De Robertis M, Copetti M, Rabitti C, Perrone G, Muda AO, Molinari F, Zanellato E, Crippa S, Caputo D, et al. Aberrant promoter methylation of beta-1,4 galactosyltransferase 1 as potential cancer-specific biomarker of colorectal tumors. Genes Chromosomes Cancer. 2012; 51:1133–1143.
19. Saldova R, Dempsey E, Pérez-Garay M, Mariño K, Watson JA, Blanco-Fernández A, Struwe WB, Harvey DJ, Madden SF, Peracaula R, McCann A, Rudd PM. 5-AZA-2′-deoxycytidine induced demethylation influences N-glycosylation of secreted glycoproteins in ovarian cancer. Epigenetics. 2011; 6:1362–1372.
20. Jacob F, Hitchins MP, Fedier A, Brennan K, Nixdorf S, Hacker NF, Ward R, Heinzelmann-Schwarz VA. Expression of GBGT1 is epigenetically regulated by DNA methylation in ovarian cancer cells. BMC molecular biology. 2014; 15:24.
21. Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome research. 2002; 12:996–1006.
22. Li L-C, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002; 18:1427–1431.
23. Li W, Cowley A, Uludag M, Gur T, McWilliam H, Squizzato S, Park YM, Buso N, Lopez R. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015; 43:W580–584.
24. Takai D, Jones PA. The CpG island searcher: a new WWW resource. In Silico Biol. 2003; 3:235–240.
25. Kelly TK, Liu Y, Lay FD, Liang G, Berman BP, Jones PA. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 2012; 22:2497–2506.
26. Anugraham M, Jacob F, Nixdorf S, Everest-Dass AV, Heinzelmann-Schwarz V, Packer NH. Specific glycosylation of membrane proteins in epithelial ovarian cancer cell lines: glycan structures reflect gene expression and DNA methylation status. Molecular & cellular proteomics : MCP. 2014; 13:2213–2232.
27. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70.
28. Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015; 15:540–555.
29. Horvat T, Zoldoš V, Lauc G. Evolutional and clinical implications of the epigenetic regulation of protein glycosylation. Clin Epigenetics. 2011; 2:425–432.
30. Saldova R, Dempsey E, Perez-Garay M, Marino K, Watson JA, Blanco-Fernandez A, Struwe WB, Harvey DJ, Madden SF, Peracaula R, McCann A, Rudd PM. 5-AZA-2′-deoxycytidine induced demethylation influences N-glycosylation of secreted glycoproteins in ovarian cancer. Epigenetics. 2011; 6.
31. Badve S, Dabbs DJ, Schnitt SJ, Baehner FL, Decker T, Eusebi V, Fox SB, Ichihara S, Jacquemier J, Lakhani SR, Palacios J, Rakha EA, Richardson AL, et al. Basal-like and triple-negative breast cancers: a critical review with an emphasis on the implications for pathologists and oncologists. Mod Pathol. 2011; 24:157–167.
32. Yoshimura M, Yoshimura M, Ihara Y, Ihara Y, Matsuzawa Y, Matsuzawa Y, Taniguchi N, Taniguchi N. Aberrant glycosylation of E-cadherin enhances cell-cell binding to suppress metastasis. J Biol Chem. 1996; 271:13811–13815.
33. Sato Y, Takahashi M, Shibukawa Y, Jain SK, Hamaoka R, Miyagawa Ji, Yaginuma Y, Honke K, Ishikawa M, Taniguchi N. Overexpression of N-acetylgluco saminyltransferase III enhances the epidermal growth factor-induced phosphorylation of ERK in HeLaS3 cells by up-regulation of the internalization rate of the receptors. J Biol Chem. 2001; 276:11956–11962.
34. Xu Q, Akama R, Isaji T, Lu Y, Hashimoto H, Kariya Y, Fukuda T, Du Y, Gu J. Wnt/beta-catenin signaling down-regulates N-acetylglucosaminyltransferase III expression: the implications of two mutually exclusive pathways for regulation. J Biol Chem. 2011; 286:4310–4318.
35. Isaji T, Gu J, Nishiuchi R, Zhao Y, Takahashi M, Miyoshi E, Honke K, Sekiguchi K, Taniguchi N. Introduction of bisecting GlcNAc into integrin alpha5beta1 reduces ligand binding and down-regulates cell adhesion and cell migration. J Biol Chem. 2004; 279:19747–19754.
36. Kelly TK, Liu Y, Lay FD, Liang G, Berman BP, Jones PA. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome research. 2012; 22:2497–2506.
37. Brehove M, Wang T, North J, Luo Y, Dreher SJ, Shimko JC, Ottesen JJ, Luger K, Poirier MG. Histone core phosphorylation regulates DNA accessibility. The Journal of biological chemistry. 2015; 290:22612–22621.
38. Jacob F, Hitchins MP, Fedier A, Brennan K, Nixdorf S, Hacker NF, Ward R, Heinzelmann-Schwarz VA. Expression of GBGT1 is epigenetically regulated by DNA methylation in ovarian cancer cells. BMC Mol Biol. 2014; 15:24.
39. Jacob F, Guertler R, Naim S, Nixdorf S, Fedier A, Hacker NF, Heinzelmann-Schwarz V. Careful selection of reference genes is required for reliable performance of RT-qPCR in human normal and cancer cell lines. PloS one. 2013; 8:e59180.
40. Kumaki Y, Oda M, Okano M. QUMA: quantification tool for methylation analysis. Nucleic Acids Res. 2008; 36:W170–175.
41. Cooper CA, Gasteiger E, Packer NH. GlycoMod—a software tool for determining glycosylation compositions from mass spectrometric data. Proteomics. 2001; 1:340–349.
42. Ceroni A, Maass K, Geyer H, Geyer R, Dell A, Haslam SM. GlycoWorkbench: a tool for the computer-assisted annotation of mass spectra of glycans. J Proteome Res. 2008; 7:1650–1659.
43. Cline MS, Craft B, Swatloski T, Goldman M, Ma S, Haussler D, Zhu J. Exploring TCGA Pan-Cancer data at the UCSC Cancer Genomics Browser. Sci Rep. 2013; 3:2652.
44. Lopez-Bigas N, Cline M, Broom B, Margolin A, Omberg L, Weinstein J, Axton M. Thread 4: Data discovery, transparency and visualization. Nature Genetics. 2013.
45. Bibikova M, Fan J-B. GoldenGate assay for DNA methylation profiling. Methods Mol Biol. 2009; 507:149–163.
46. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1.