INTRODUCTION
The complex network of DNA repair system in mammals counteracts genome insult and maintains genomic stability. DNA repair failure increases mutations frequency and affects cell-cycle regulation, promoting tumorigenesis; in fact, inactivating mutations in DNA repair pathways are frequent in cancer. On the other hand, DNA repair defects provide new therapeutic opportunities to treat cancer through DNA-damage-inducing radiation and chemotherapies. Identification of synthetic interactions, i.e., negative genetic combination of mutations in two or more genes, leading to severe slow growth or lethality compared to single mutants, has grown in popularity in the DNA damage field, with the finding that poly(ADP-ribose) polymerase (Parp-1) inhibitors show synthetically lethal effects in homologous recombination (HR) defective tumors [1].
PolyADP-ribosylation acts in DNA-damage repair response and maintenance mechanisms of genomic stability. Several DNA repair pathways, including base-excision repair and double strand break (DSB) repair, involve Parp functions. Inhibition of Parp-1 results in an increase in single strand breaks (SSBs) that then collapse replication forks into toxic one-ended DSBs [1,2], which are substrates for HR [3,4]. In addition, through enzyme-dependent chromatin remodeling and enzyme-independent motif recognition, Parp-1 also plays important roles in regulating gene expression [5]. Furthermore, Parp-1 binding motifs may be readily found in promoter elements of DNA repair genes, linking DNA repair and transcription functions of Parp-1 [5]. The critical role of Parp-1 in maintenance of genomic stability is reflected in its frequent upregulation in cancer [6,7], and by hypersensitivity of Parp-1 null animals toward the mutagenic effect of DNA damaging agents [8].
The Rad54 gene encodes a dsDNA-dependent ATPase of the Swi2/Snf2 family [9], which interacts directly with Rad51 [10,11], and stimulates its DNA exchange activity [12]. Rad54 promotes chromatin remodeling, Rad51 displacement from double-stranded DNA, binds Holliday junctions and drives their branch migration [13]. Although disruption of other genes involved in HR leads to embryonic lethality, adult Rad54−/− mice are viable and fertile, providing a valuable model to study the effects of attenuated HR. Interestingly, several point mutations in conserved regions of the Rad54 gene family have been found in primary tumors (e.g., breast and colon carcinomas and lymphomas) [14,15], and approximately half of epithelial ovarian cancers have alterations in genes regulating HR repair [16].
Most DNA repair pathways are complex, involving many proteins working in discrete consecutive steps. Therefore, their balanced and coordinated expression is important to avoid erroneous repair that might result from excessive base removal and DNA cleavage. In addition, as DNA damage is a likely factor in promoting central nervous system (CNS) pathophysiology, DNA repair may be an important mechanism for maintenance of normal physiological function. In this study, to investigate the functional relationship between the HR factor Rad54 and Parp-1 in development and oncogenesis in vivo, we introduced single or combined Rad54 and Parp-1 inactivating mutations in a well characterized cancer model, the Ptc1 heterozygous mice. Mice in which one copy the Ptc1 gene has been inactivated (Ptc1+/-) are developmentally nearly normal, but show a marked predisposition to tumor development, including medulloblastoma (MB), a frequent pediatric malignant tumor of the cerebellum [17,18]. Our previous work showed that neonatal irradiation dramatically increases the frequency of MB [19,20]. We also showed that combined loss of Ptc1 and Rad54 or of Ptc1 and Parp-1 increased radiation induced MB [21,22]. Here, we analyzed the effects of combined loss of Rad54 and Parp-1 in the Ptc1 mouse model of spontaneous and radiation-induced cancer, highlighting novel synthetic lethal interactions during development and tumorigenesis, and dissecting the underlying cellular and molecular mechanisms.
RESULTS
Survival and oncogenesis in crosses between Rad54, Parp-1 and Ptc1 mutant mice
Defects in DNA damage signaling are frequently associated with neurodegeneration, neurodevelopmental disease and brain tumors. We therefore sought to determine the oncogenic potential of combined genetic disruption of Rad54 and Parp-1 in the CNS by knocking out these genes in Ptc1+/- mice. To this aim, Rad54+/-/Parp-1+/-/Ptc1+/- and Rad54+/-/Parp-1+/-/Ptc1+/+ mice were intercrossed, and the progeny genotyped. For simplification, wt genotypes are hereafter omitted from the label, unless differently specified. Mutants for Rad54 (Rad54-/-/Parp-1+/-/Ptc1+/-) or Parp-1 (Rad54+/-/Parp-1-/-/Ptc1+/-) were viable. Irrespective of Ptc1 genotype, no mice with compound Rad54/Parp-1 inactivation were found at weaning, showing strong synthetic lethal interaction between Rad54 and Parp-1 genes in vivo, in agreement with existing in vitro data. To look for haploinsufficient interactions, compound mutant mice with varying gene dosage of Rad54 and Parp-1 were analyzed for survival and cerebellum tumorigenesis, with or without irradiation. Because only Ptc1+/- mice are prone to MB, the effects of Rad54 and Parp-1 inactivation was evaluated only in mice with Ptc1+/- genotype.
Lack of Parp-1, or Rad54 per se affected lifespan of Ptc1+/- mice by significantly shortening the median survival time from 64 to 14 (P ˂ 0.0001) or 45 weeks (P = 0.0206), respectively (Figure 1A and 1C). However, loss of Parp-1 function had more severe effects on survival compared to Rad54 loss (i.e., 14 vs 45 weeks; P = 0.0002) (Figure 1A), suggesting a preferential role for Parp-1 in resolution of spontaneous DNA damage compared to Rad54. Irradiation of Ptc1+/- mice or Ptc1/Rad54 compound mutants (Rad54-/-/Ptc1+/-) with 1 Gy of x-rays caused a sharp reduction of lifespan relative to controls (17 vs 64 weeks P ˂ 0.0001; 13 vs 45 weeks; P = 0.0002; Figure 1A-1C), with median survival reduced approximately 3-fold. Notably, life shortening was not affected by Rad54 status, in agreement with findings that Rad54-/- mice are not hypersensitive to radiation [23]. Moreover, irradiated Ptc1/Parp-1 compound mutants (Parp-1-/-/Ptc1+/-) did not show further survival reduction compared to controls (15 vs 14 weeks; P = 0.862; Figure 1A-1C). The lack of modifying effects of Parp-1 on Ptc1-associated radiosensitivity is consistent with previous reports from our laboratory [22].
Coherent with survival reduction, genetic disruption of Parp-1 significantly increased spontaneous MB incidence in Ptc1+/- mice (50% vs 14% in Parp-1-/-/Ptc1+/- and Ptc1+/- mice, respectively; P = 0.0038) or in Ptc1+/- mice lacking Rad54 (i.e., the viable genotypes: Rad54+/-/Parp-1-/-/Ptc1+/- and Rad54-/-/Ptc1+/-, 44% vs 15%, respectively; P = 0.0274) (Figure 1D-1F). Irradiation of Ptc1+/- (Ptc1+/-) or Rad54-/- mice (Rad54-/-/Ptc1+/-) caused significantly increase MB incidence compared with unirradiated mice, both in wt Parp-1 background [Ptc1+/- (82% vs 14%; P = 0.0001); Rad54-/-/Ptc1+/- (84% vs 15%; P = 0.0001)] and in Parp-1 heterozygotes (Parp-1+/-/Ptc1+/- 56% vs 30%; P = 0.0447) (Figure 1D-1F). In contrast, irradiation of Parp-1-/-/Ptc1+/- mice caused a non-statistical increase of their already high MB incidence (75% vs 50%; P = 0.1717). This difference became significant only in mice with concurrent inactivation of one copy of Rad54 [(Rad54+/-/Parp-1-/-/Ptc1+/- mice) (79% vs 44%; P = 0.0378)] (Figure 1E and 1F). Notably, in a null Rad54 background deletion of one Parp-1 allele caused significantly lower radiation-induced MB tumorigenesis compared with Parp-1 wt mice [Rad54-/-/Parp-1+/-/Ptc1+/- (56%) vs Rad54-/-/Ptc1+/- mice (84%), P = 0.0439] (Figure 1E and 1F), suggesting a lethal haploinsufficient interaction between Rad54 and Parp-1 in the context of radiation oncogenesis. In all groups, irradiation increased the incidence of MB, an early-onset tumor. This was reflected in decreased incidence of late occurring tumors, including sarcomas and other tumors (Figure 1F), with exception of Parp-1-/-/Ptc1+/- mice, in which MB mortality was already high and did not significantly increase with irradiation.
Loss of the normal Ptc1 allele is the causative event in MB development in Ptc1+/- mice [24]. To test if single or combined Rad54 and Parp-1 deficiency alters the typical chr-13 LOH pattern in MB, we performed microsatellite analysis with a set of markers spanning the length of chr-13, where Ptc1 gene is located (Figure S1). Within the sample size, no obvious differences were observed in Ptc1 LOH pattern in single or compound Rad54 and Parp-1 mutants.
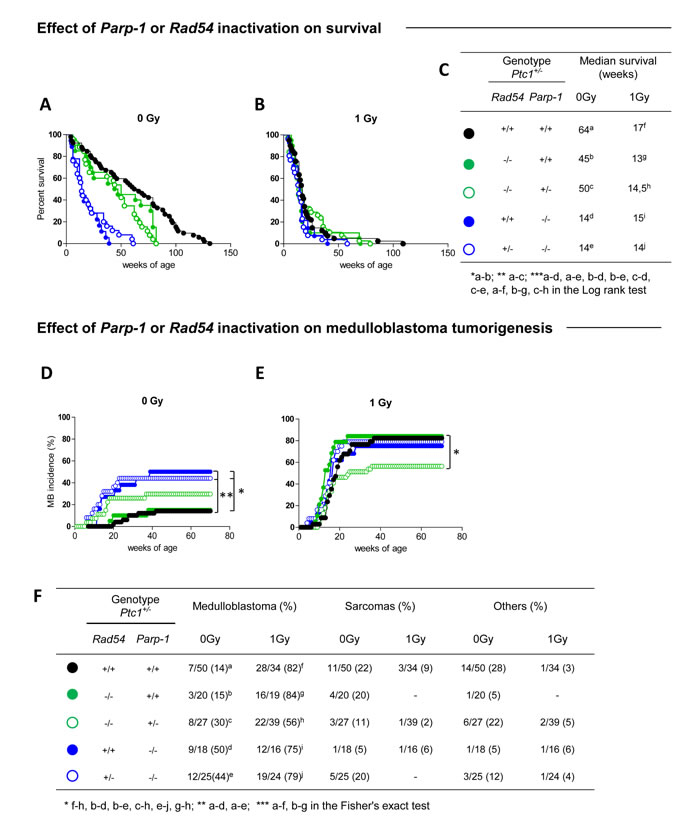
Figure 1: Effect of Rad54 and Parp-1 inactivation on survival and tumor development. (A) Survival curves of unirradiated Ptc1+/- , Rad54-/-/Ptc1+/-, Rad54-/-/Parp-1+/-/Ptc1+/-, Parp-1-/-/Ptc1+/- and Rad54+/-/Parp-1-/-/Ptc1+/- mice, showing significant lifespan shortening after inactivation of Parp-1. (B) Survival curves of Ptc1+/-, Rad54-/-/Ptc1+/-, Rad54-/-/Parp-1+/-/Ptc1+/-, Parp-1-/-/Ptc1+/- and Rad54+/-/Parp-1-/-/Ptc1+/- mice irradiated with 1Gy at P1, all showing significant lifespan shortening compared to control mice with exception of Parp-1 null mice (Rad54+/+ or +/-/Parp-1-/-/Ptc1+/-). (C) Median survival of unirradiated and irradiated mice. (D) Effect of Rad54 and Parp-1 inactivation on spontaneous and (E) radiation-induced medulloblastoma tumorigenesis. (F) Percent incidence of medulloblastoma, sarcoma and other tumors for each mouse group. *P ≤ 0.05; **P ≤ 0.005; *** P ≤ 0.0001.
Combined inactivation of Rad54 and Parp-1 causes growth retardation and perinatal lethality
The complete lack of Rad54-/-/Parp-1-/- null mice in our triple cross at weaning age prompted us to investigate the effect of this genetic interaction at earlier developmental times. Analysis of the genotype distribution in the offspring at postnatal day 1 (P1) revealed that, of 764 newborns derived from heterozygous mating, only 10 and 13 in wt and Ptc1+/- genetic backgrounds, respectively, were Rad54-/-/Parp-1-/- compound mutants, far below the 24 mice expected based on a Mendelian inheritance ratio of 25%, although this trend reached statistical significance only in Ptc1+/+ mice (Table 1). In addition, double knockout Rad54-/-/Parp-1-/- pups at P1 and P7 showed significantly smaller body size compared to relative littermates (Figure 2A and 2C). In fact, inactivation of both genes significantly reduced body weight at P1 (by 34% in wt and 17% in Ptc1+/- genotypes) and P7 (by 37% in wt and 20% in Ptc1+/- genotypes), showing that combined loss of Rad54 and Parp-1 causes severe growth retardation (Figure 2A-2D). Moreover, 45% and 29% of the double mutants on Ptc1+/- and wt background, respectively, dyed during the first fortnight of life and none survived to weaning age (data not shown).
Table 1: Genotype distribution in the offspring of the intercross Rad54+/-/Parp-1+/-Ptc1+/+ x Rad54+/-/Parp-1+/-/Ptc1+/- at birth.
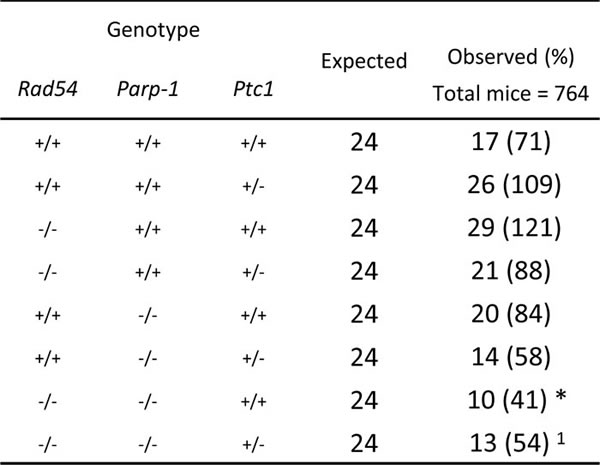
The table represents expected and observed genotype distribution in the offspring at birth. Compound mutants were born at numbers below the predicted Mendelian inheritance ratio. Mice heterozygous for Rad54 and/or Parp-1 are not shown. * P ≤ 0,05; 1P = 0,0948 Fisher's exact test.
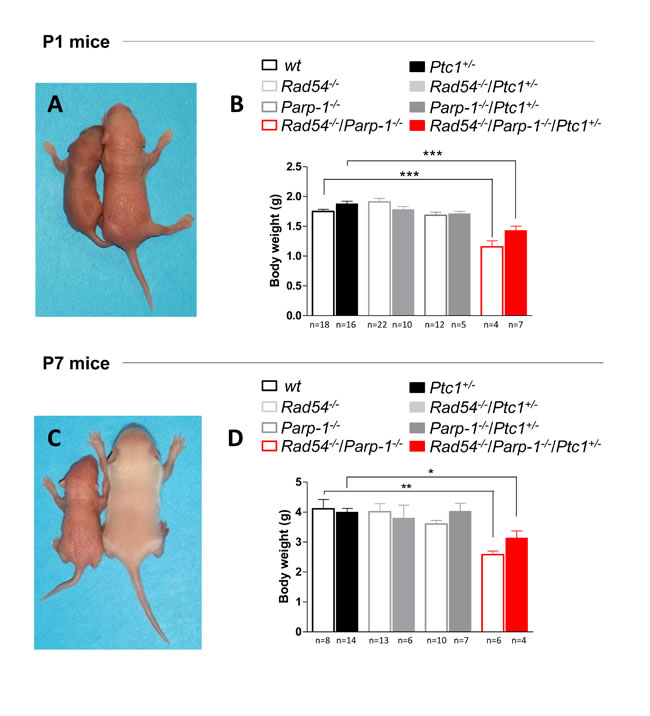
Figure 2: Effect of combined inactivation of Rad54 and Parp-1 on mouse development. Left in (A) and (C) Rad54-/-/Parp-1-/- mice exhibited reduced size compared to wt and Ptc1+/- littermates (right) at (A) postnatal day 1 and (C) 7. Double knockout Rad54-/-/Parp-1-/- mice showed significantly reduced body weight at (B) postnatal day 1 and (D) 7 compared wt and Ptc1+/- littermates, demonstrating that combined loss of Rad54 and Parp-1 causes severe growth retardation. The number of mice used per test is indicated in the graph (n). *P ≤ 0.05; **P ≤ 0.005; *** P ≤ 0.0001.
Effects of combined inactivation of Rad54 and Parp-1 on cerebellum morphogenesis
To further clarify the underlying mechanisms of synthetic lethal interactions of Rad54 and Parp-1 perturbation, we focused on the cerebellum, from which MB originates. The well-characterized developmental processes and stereotypical foliation pattern make this organ particularly amenable to the study of factors affecting development, leading to abnormal morphology or function. By measuring the cross-sectional area of developing cerebellum at P1 and P7 we show that, in line with generalized growth delay, significant decrease in size was observed in cerebella from Rad54-/-/Parp-1-/- mutants at P1 and P7 relative to Ptc1+/+ mice (by 36% at P1, P = 0.0035, and 31% at P7, P = 0.0078; Figure 3). Similarly decreased cerebellum area was observed in double null mice with Ptc1+/- genotype (by 34% at P1, P = 0.0007 and 43% at P7, P = 0.0012) (Figure 3A-3F). In keeping with growth delay, cerebella from Rad54-/-/Parp-1-/-/Ptc1+/- mice showed shallow principal fissures at P1 relative to Ptc1+/- mice (asterisks in Figure 3C). In addition, delayed lobularization was evident at P7 (Figure 3E and 3F).
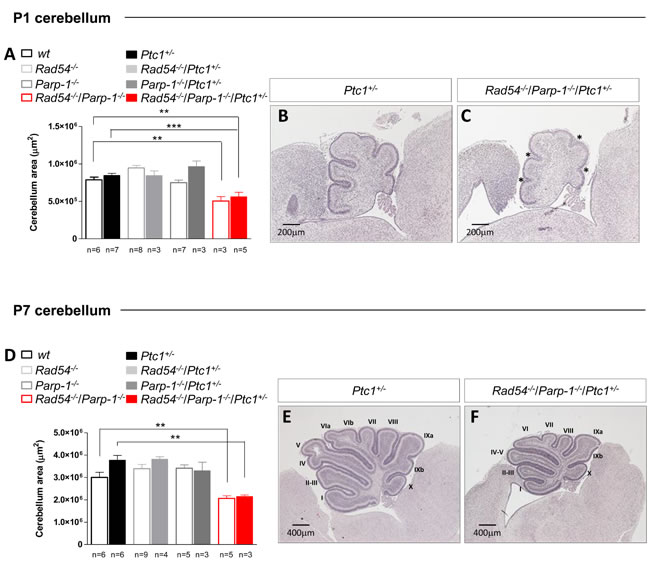
Figure 3: Effect of combined inactivation of Rad54 and Parp-1 in the developing cerebellum. Rad54-/-/Parp-1-/- double mutant mice show significant reduction of cerebellum area compared to wt and Ptc1+/- mice both at (A) P1 and (D) P7. (B and E) H & E stained sagittal sections of Ptc1+/- and (C and F) Rad54-/-/Parp-1-/-/Ptc1+/- cerebellum at P1 and P7. The number of mice used per test is indicated in the graph (n). *P ≤ 0.05; **P ≤ 0.005; *** P ≤ 0.0001.
Proliferation, DNA damage and cell death in CGNPs of cerebellum in Rad54 and Parp-1 double mutants
We next analyzed the cellular mechanisms underlying synthetic lethal interactions between Rad54 and Parp-1 by focusing on the cerebellum of Ptc1+/- mutants. The neonatal cerebellum undergoes rapid postnatal development, occurring through a burst of proliferation of granule cell precursors (GCPs) in the external granule layer (EGL) on the cerebellar surface. Notably, highly proliferating cells are particularly susceptible to endogenous and exogenous DNA damage, due to the presence of numerous replication forks that may stall and eventually undergo DSBs after collision with DNA damage [25,26]. This makes this cellular population suitable for investigating DNA repair-related mechanisms responsible for the growth delay observed. Quantification of Ki-67 staining in the EGL revealed a high proliferation rate in Parp-1-/-/Ptc1+/- mice (Figure 4A-4E), which is in keeping with the higher spontaneous MB tumorigenesis detected in Parp-1-/- mice. Similarly high growth rates were not observed in Rad54-/-/Parp-1-/-/Ptc1+/- mice, suggesting that persistent SSBs formation due to Parp-1 inhibition, promoting collapse of replication forks and DSBs which would normally be repaired by HR, cannot be repaired effectively in the absence of functional Rad54.
We therefore evaluated whether concomitant inactivation of Rad54 and Parp-1 enhances DNA damage and apoptosis in the neural precursor population of the EGL of double- compared to single-mutants or wt mice. To this aim, P1 brains were immunostained with a DSB (γ-H2AX), or an apoptotic marker (cleaved caspase-3), and the staining in the EGL was quantified. Consistent with its recognized role in resolution of SSB and stalled replication forks, inactivation of Parp-1, but not Rad54, resulted in a significant 3-fold increase of spontaneous DSBs compared to Ptc1+/- mice (P = 0.0013). A 4-fold increase (P = 0.0010) was also detected in Rad54-/-/Parp-1-/-/Ptc1+/- mutants (Figure 4F-4J). However, despite similar amount of DSBs induced by inactivation of Parp-1 and by combined inactivation of Parp-1 and Rad54, we detected large differences in positivity for cleaved caspase-3 among the different mutants (Figure 4K-4O). Mice with combined inactivation of Rad54 and Parp-1 (Rad54-/-/Parp-1-/-/Ptc1+/-) showed significantly increased staining for cleaved caspase-3 expression compared to Rad54-/- (Rad54-/-/Ptc1+/-; 15.3-fold, P = 0.007), Parp-1-/- (Parp-1-/-/Ptc1+/-; 3.9-fold, P = 0.020) or Ptc1+/- (Ptc1+/-; 9.3-fold, P = 0.003) mice. These findings confirm that Parp-1 inactivation causes an excess of spontaneous DSBs in neural cells, which can be partly repaired if Rad54 is active, but result in high apoptotic levels in GCPs with concurrent Rad54 inactivation.
In the neonatal cerebellum, the proliferation phase is followed by differentiation processes, consisting in cell cycle exit and inward migration of postmitotic GCPs to the internal granule layer (IGL), which causes progressive EGL disappearance and formation of the IGL of mature granule neurons. To evaluate the effects of combined inactivation of Rad54 and Parp-1 on differentiation processes, we measured the areas of the EGL and IGL in the cerebellum of Ptc1+/- mice with single or combined inactivation of Rad54 or Parp-1 at P7, and calculated their ratio. Despite drastic decrease in cerebellum size, no modification of the EGL/IGL ratio was detected in mice with combined inactivation of Rad54 and Parp-1 relative to other genotypes, indicating no overt alterations in the balance between proliferation (EGL) and differentiation (IGL) in the cerebellum caused by single or combined inactivation of the genes under investigation (Figure 4P). However, quantification of the density of neurons labeled by the NeuN marker of mature neurons in fixed regions of the IGL (crest number VIII, red square in Figure 4R and 4S), revealed a significant decrease in the density of NeuN+ mature neurons in Parp-1-/-/Ptc1+/- (43.5%, P = 0.0126) and Rad54-/-/Parp-1-/-/Ptc1+/- mice (45.7%, P = 0.0042), suggestive of depletion of granule neurons (Figure 4Q and 4S).
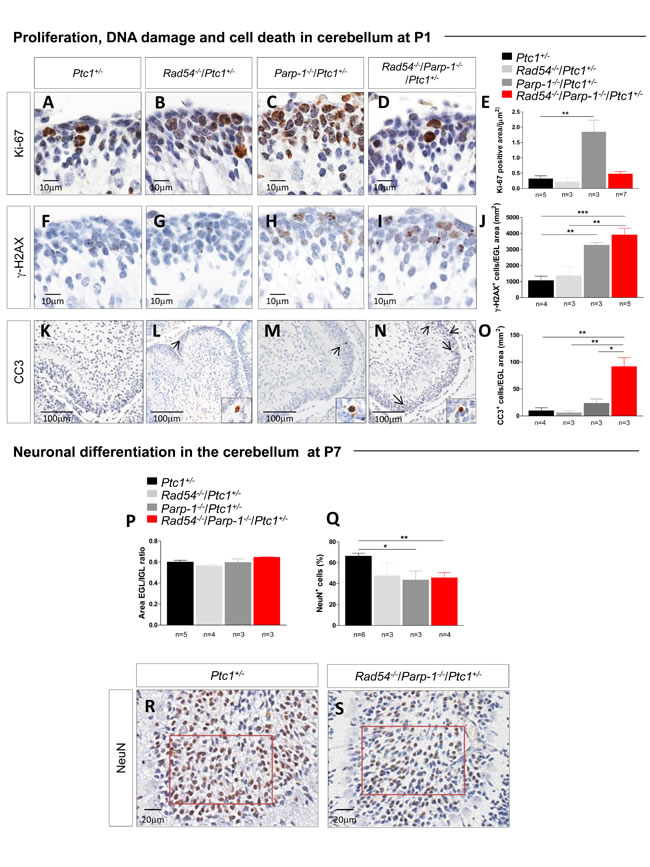
Figure 4: Evaluation of proliferation, DNA damage and cell death in neuronal precursors of the EGL at P1 and differentiation in mature granule neurons in the IGL at P7. (A-E) Immunohistochemical analyses for Ki-67 showing significant increased expression in Parp-1-/-/Ptc1+/- compared to Ptc1+/- mice. (F-J) Immunohistochemical analyses for γ-H2AX showing significantly increased expression in Parp-1-/-/Ptc1+/- and Rad54-/-/Parp-1-/-/Ptc1+/- compared to Ptc1+/- mice. (K-O) Immunohistochemical analyses for cleaved caspase-3 (CC3) showing significantly increased expression in Rad54-/-/Parp-1-/-/Ptc1+/- mice compared to Rad54-/-/Ptc1+/-, Parp-1-/-/Ptc1+/- or Ptc1+/- mutant mice. (P) The ratio between EGL and IGL area was similar in mice of all genotypes. (Q) Significant decrease in number of mature neurons expressing NeuN in Parp-1-/-/Ptc1+/- and Rad54-/-/Parp-1-/-/Ptc1+/- mutants compared to Ptc1+/- mice. (R and S) Expression level of NeuN was determined in a fixed area (in red) from folia VIII. The number of mice used per test is indicated in the graph (n). *P ≤ 0.05; **P ≤ 0.005; *** P ≤ 0.0001.
Synthetic lethality affects the expression of DNA repair related gene
The robust synthetic lethal effect of combined Rad54 and Parp-1 inactivating mutations led us to investigate the underlying molecular mechanisms by addressing the modification of the expression profile of 84 genes with established roles in DNA damage signaling. We carried out this analysis in the P1 cerebellum of Ptc1+/- mice with single or combined inactivation of Rad54 and/or Parp-1 (Rad54-/-/Ptc1+/-, Parp-1-/-/Ptc1+/- and Rad54-/-/Parp-1-/-/Ptc1+/-) and results were compared with those from Ptc1+/- mice using a pathway-based PCR expression array. In Rad54-/-/Ptc1+/- mice 14 genes were significantly downregulated (16.7%) and 8 upregulated (9.5%); in Parp-1-/-/Ptc1+/- mice, 22 genes were significantly downregulated (26%) and 6 (7%) upregulated; in Rad54-/-/Parp-1-/-/Ptc1+/- mice, 10 genes (12%) were downregulated and 16 (19%) upregulated (S1Table). The magnitude of many of these changes was small. However, as DNA repair genes are expressed at basal level in mammals, detection of small changes does not necessarily mean a negligible biological significance, but rather that even slight deregulation may significantly modify DNA repair capacity. We also performed a hierarchical clustering analysis that produced different groups of genes (colored bars, left side) based on similar fold-change patterns among the three genotypes (Rad54-/-/Ptc1+/-, Parp-1-/-/Ptc1+/-, Rad54-/-/Parp-1-/-/Ptc1+/- mice) (Figure 5A). In particular, the violet module (lowest bar) shows the most upregulated genes in Rad54-/-/Parp-1-/-/Ptc1+/- mice (i.e., Exo1, P = 0.031; Pttg1, P = 0.001); in the gray module are listed the genes downregulated in Rad54-/-/Ptc1+/- and Parp-1-/-/Ptc1+/- mutants that are upregulated in Rad54-/-/Parp-1-/-/Ptc1+/- mice, of which Xpa was statistically significant (P = 0.033); instead, in the red module, the synergistic effects of combined Rad54 and Parp-1 mutations lead to downregulation of several genes (i.e., Atm, P = 0.031; Atr, P = 0.008; Atrx, P = 0.006; Parp-1, P = 0.001; Prkdc, P = 0.029; Rad50, P = 0.028; Wrn, P = 0.016) (Figure 5A and Table S1).
Pathway analysis of all significantly deregulated genes was performed using SPIA algorithm. The signaling pathway showing a significant perturbation in Rad54-/-/Parp-1-/-/Ptc1+/- mice was the “Cell Cycle” pathway, including 10 genes (i.e. Atm, Cdc25c, Cdkn1a, Pcna, Prkdc, Rad21, Gadd45g, Smc1a, Atr, Pttg1) with fold changes and topological distribution suggesting pathway inhibition. Further analysis of these genes, performed with the Cytoscape gene ontology tool Cluego, showed a substantial overlap with the p53 signaling pathway (Figure 5B).
We validated Atm and Prkdc (DNA-PKcs), by RT-PCR confirming a significantly decreased expression in Rad54 and Parp-1 compound mutants (Rad54-/-/Parp-1-/-/Ptc1+/-) compared to single mutants (Figure 5C) and verified the decreased expression levels of ATM and DNA-PKcs by immunoblotting (Figure 5D).
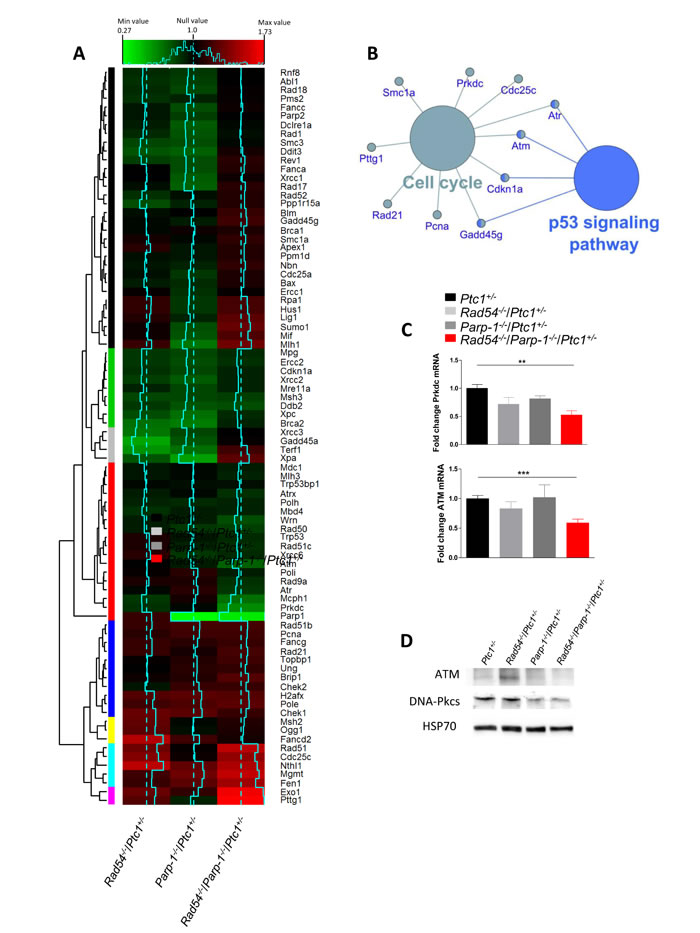
Figure 5: Bioinformatic analysis of expression profiles of DNA repair-related genes in P1 cerebella from Rad54-/-/Ptc1+/-, Parp-1-/-/Ptc1+/-, Rad54-/-/Parp-1-/-/Ptc1+/- and Ptc1+/- mice. (A) Heat map, generated from RT2 Profiler PCR Arrays Mouse DNA Damage, representing the fold-change variation in the three genotypes (solid blue lines) versus the Ptc1+/- mice (dotted blue lines). Relative hierarchical clustered groups represented by colored bars on left side. (B) Gene Ontology network generated by analysis of the most significantly perturbed KEGG pathways in the Rad54-/-/Parp-1-/-/Ptc1+/- mice when compared to the Ptc1+/- mice. (C) SYBR Green real-time PCR validation of ATM and Prkdc (DNA-PKcs) genes. (D) Immunoblot analysis of ATM and DNA-PKcs proteins with relative HSP70 as loading control. *P ≤ 0.05; **P ≤ 0.005; *** P ≤ 0.0001.
p53/p21-mediated senescence as underlying mechanism for the lethal phenotype of Rad54-/-/Parp-1-/- compound mutants
Based on results of bioinformatic analysis, we analyzed p53 expression in protein extracts from P1-cerebellum by immunoblotting. Although no significant differences were detected between Rad54 and Parp-1 single and compound mutants in phosphorylation of p53 at Ser18 [27], we observed a significant increase in total p53 expression (3-fold) only in compound mutants, indicating marked p53 activation (Figure 6A), which is consistent with persistent genetic damage. We also show that p53 stabilization did not occur at transcriptional level, as no changes in the expression of p53 were detected in the array (Figure 5A and Table S1), suggesting, instead, posttranslational modification. Immunoblotting for p21 showed a trend towards increased expression that, however, was not significant (Figure 6A).
To evaluate the effect of combined Rad54 and Parp-1 mutations at organismal level, we established mouse embryonic fibroblasts (MEFs) from Rad54-/-/Parp-1-/-/Ptc1+/- and Ptc1+/- mice. Immunoblot analysis of p53/p21 signaling pathway in MEFs of different genotypes showed significantly increased expression of p53-P(ser18), p53 and p21 in Rad54-/-/Parp-1-/-/Ptc1+/- mutants compared to Ptc1+/- MEFs (Figure 6B). Moreover, MEFs from Rad54-/-/Parp-1-/-/Ptc1+/- mice showed decreased cell growth potential compared to those from Ptc1+/- mice. The difference in growth rate become significant at 72 and 96 h after seeding, with a 1.5-2 fold reduction in cell numbers and an increase in doubling times (23 vs 35 h) in MEFs from Rad54-/-/Parp-1-/-/Ptc1+/- compared to those from Ptc1+/- mice (Figure 6C). We also detected a significantly greater fraction of positive cells after staining with Senescence-Associated β-Galactosidase (SA-β-GAL) (1.6-fold; P = 0.048), a biomarker of senescent cells, accompanied by senescence-associated morphological cellular changes (i.e., enlargement and flattening) (Figure 6E and 6F), suggesting accelerated acquisition of senescence in Rad54-/-/Parp-1-/-/Ptc1+/- compared to Ptc1+/- MEFs.
Although identification of senescent cells is challenging, especially in vivo, loss of replicative markers such as Ki-67 and sustained DNA damage are often considered as bona fide markers of senescence. We immunostained several tissues (thymus, lung, kidney, liver, intestine, skin) from Rad54-/-/Parp-1-/-/Ptc1+/- and Ptc1+/- mice at P1 with Ki-67 and γ-H2AX. Compared to Ptc1+/- mice, we observed a general decrease in proliferation and increase in DNA damage in all tissues from Rad54-/-/Parp-1-/-/Ptc1+/- mice, particularly in thymus, intestine and kidneys (Figure 6G-6R). Instead, immunostaining with caspase-3 did not reveal obvious differences in apoptosis (data not shown). Altogether, these data strongly support the involvement of p53/p21-mediated accelerated senescence in growth delay of Rad54-/-/Parp-1-/- compound mutants, culminating in a lethal perinatal phenotype.
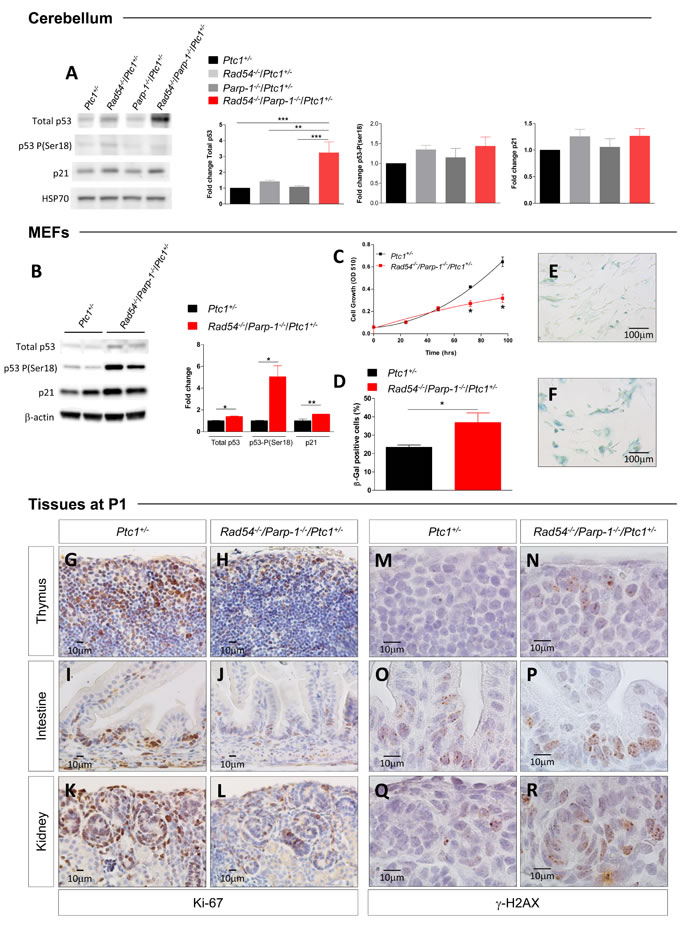
Figure 6: Evaluation of biomarkers of senescence. (A) Immunoblot and densitometric analysis of p53/p21 signaling [total p53, p53-P(ser18) and p21] in cerebellum of Ptc1+/-, Rad54-/-/Ptc1+/-, Parp-1-/-/Ptc1+/-, and Rad54-/-/Parp-1-/-/Ptc1+/- mice at P1, and (B) in MEFs from Ptc1+/- and Rad54-/-/Parp-1-/-/Ptc1+/- mice (two independent cell lines per genotype); β-actin and HSP70 were used as loading controls. (C) Growth kinetics and (D) quantification of SA-β-GAL staining in MEFs from Ptc1+/- and Rad54-/-/Parp-1-/-/Ptc1+/- mouse embryos. (E) Representative images of SA-β-GAL staining in MEFs from Ptc1+/-, and (F) and Rad54-/-/Parp-1-/-/Ptc1+/- mouse embryos. (G-L) Proliferation (Ki67) and (M-R) DNA damage (γ-H2AX) in different tissues from Ptc1+/- and Rad54-/-/Parp-1-/-/Ptc1+/- neonatal mice at P1. The number of mice used per test is indicated in the graph (n). *P ≤ 0.05; **P ≤ 0.005; *** P ≤ 0.0001.
DISCUSSION
The DNA repair system functions as a critical tumor suppressor network to preserve the integrity of the genome preventing malignancy, and DNA repair defects are frequently encountered in human cancer. Therefore, gaining insights into the mechanism of action of DNA repair genes is pivotal to development of novel tumor-specific treatment strategies targeting deregulated DNA repair [28]. A major obstacle to achieving synthetic lethal therapies is a lack of insight into the phenomenon in cancer cells, and insufficient knowledge of the molecular mechanisms influencing synthetic lethal interactions.
We earlier investigated in vivo genetic interactions between Parp-1 and Ptc1, and found that combined inactivation of the two genes is not critically important for normal development and survival of the compound mutant mice; however, increased genetic instability caused by abrogation of Parp-1 was able to promote MB formation in Ptc1+/- mice by fostering accumulation of genetic defects and facilitation of loss of the remaining normal Ptc1 allele [22]. Despite the critical role of the HR system for embryonic development, attenuated HR by deletion of Rad54 also did not result in lethality in Ptc1+/- mice, implying redundancy in function of proteins involved in HR; however, in previous work, by exposing double Rad54/Ptc1 mutants to 2 Gy, we detected significantly increased MB tumorigenesis, possibly through the generation of viable but genetically rearranged neural cell precursors due to repair of DNA lesions by error-prone mutagenic pathways [21]. An analogous increase was not seen here, likely due to the lower dose used and relative radioresistance of Rad54-/- mice [23].
Former in vitro studies have shown that Parp-1 inhibitors induce synthetic lethality in cells that are defective in HR [1,2,29]. An extensive body of literature has since documented the impact of Parp inhibitors on synthetic lethality in cells that are defective in HR due to loss of HR-associated proteins, and their use as single-agent therapies acting through the concept of synthetic lethality [30]. The results shown here demonstrate for the first time that Parp-1 and Rad54 are genetically linked and that they share overlapping functions that are essential in mouse development. We observed not only that the combination is active in vivo, but could also clarify that Parp-1 is not strictly necessary for HR during early embryogenesis, as combined deletions of Parp-1 and Rad54 caused significantly reduced birth rate and growth retardation - resulting in early death of Rad54-/-/Parp-1-/- pups - not full embryonic lethality.
Since evaluation of the effects of simultaneous Rad54 and Parp-1 inactivation on MB development was prevented by the lack of viable compound mutants, we investigated the inactivation of DNA repair genes in the context of Ptc1 tumor suppression by examining mice with complete abrogation of either Rad54 or Parp-1 function, in combination with heterozygosis of Parp-1 or Rad54, respectively. Attenuation of Rad54 in the context of Parp-1 abrogation had synthetic lethal effects in dosage-independent manner. In contrast, inactivation of one copy of Parp-1 in the setting of Rad54 abrogation had two distinct outcomes depending on treatment, i.e., no effect on spontaneous tumor development, or decreased induction of MB induced by radiation. A further increase in Parp-1 inactivation (i.e., both copies) caused a shift to synthetic lethality (i.e., loss of viable progeny), demonstrating that combined defects in Rad54 and Parp-1 cause synthetic lethal interactions in a Parp-1-gene dosage dependent manner, and highlighting the delicate balance that must be hit when targeting essential pathways.
Important to our understanding of the underlying mechanisms of this novel genetic link, immunohistochemical analysis revealed a trend to high degree of granule cell proliferation in the EGL of Parp-1 null mice (Parp-1-/-/Ptc1+/-), which decreased significantly in age-matched double mutants (Rad54-/-/Parp-1-/-/Ptc1+/-). This was accompanied by substantial delay in neuronal differentiation, as shown by sparse NeuN staining and limited presence of committed GCPs in both groups. Given the concurrent high levels of spontaneous DSBs in both Parp-1 null mutants (Parp-1-/-/Ptc1+/- and Rad54-/-/Parp-1-/-/Ptc1+/-), the low apoptotic levels observed only in Parp-1-/-/Ptc1+/- correlate well with the high spontaneous rate of MB in these mice, and suggest that the cells can survive with a mutation in either repair pathways, not both, which causes cells to die.
As several DNA repair deficiency syndromes (e.g. ataxia telangiectasia, and spinocerebellar ataxia with axonal neuropathy1) target the cerebellum [31], we set up to investigate how alternative repair pathways compensate the impairment of predominant DNA repair mechanisms to maintain cerebellum integrity. We compared gene expression changes in DNA repair-related genes in Rad54 and Parp-1 single (Rad54-/-/Ptc1+/-; Parp-1-/-/Ptc1+/-) and combined mutants (Rad54-/-/Parp-1-/-/Ptc1+/-) versus Ptc1+/- mice. We detected strong downregulation of Prkdc (DNA-PKcs), which was unexpected as DNA-PKcs activation has been invoked to explain the synthetic lethal effects of Parp-1 inhibitors in HR-defective cells [32]. DNA-PKcs downregulation was paralleled by marked downregulation of Atm. DNA-PKcs and Atm are both members of the phosphatidylinositol 3-kinase (PI3K)-related protein kinase superfamily and act in tandem by important cross-talk [33,34]. Notably, combined loss of Atm and DNA-PKcs cause embryonic lethality in mice [35,36], suggesting that their downregulation may be central to the perinatal lethality of Rad54-/-/Parp-1-/- mutants. PI3K and Parp-1 inhibitors has proven effective in treating Brca1-related breast cancer in vivo [37].
Driven by the results of the bioinformatic analysis converging on p53 signaling, we investigated the expression of p53 protein in P1-cerebellum and MEFs, detecting significant accumulation of p53 protein in both. Because p53 phosphorylation is classically considered as the first crucial step of p53 stabilization, driven by several kinases, including ATM and DNA-PKcs, we analyzed phosphorylation of p53 at Ser18 [27] in P1 cerebella and did not detect significant differences among genotypes, despite accumulation of p53 only in double mutants. However, it is known that multiple phosphorylation sites exist, acting cooperatively for stimulating p53 activity [38]. Also, phosphorylation may not be a universal requirement for p53 stabilization, especially in vivo, as several studies have established, shifting attention on Mdm2 regulation of p53 [39]. All these factors, along with differences between in vitro and in vivo conditions, may concur to explain the differences in the expression pattern of p53/p53 Ser18 observed between MEFs and cerebellum.
Our results of p53 expression in P1-cerebellum and MEFs are in line with the fact that deficiency in several DNA repair genes such as DNA Lig4, XRCC4 and Rad51 cause increased level of genotoxic stress, triggering a p53-dependent DNA damage response leading to embryonic lethality [40-42]. More generally, mutations in over 150 cell-essential genes from different pathways result in recurrent phenotypes, which involve neural apoptosis and developmental delay in zebrafish and share p53 upregulation, suggesting that p53 might function as a developmental checkpoint triggered by various stressors, including genetic defects [43]. In agreement, mice with constitutively active p53 are generally not viable, due to inhibition of proliferation - via cell-cycle arrest, quiescence, senescence and differentiation - and/or induction of apoptosis [44]. The nature and intensity of damage, as well as, the level of p53 expression and its interaction with specific proteins, may determine the choice among the different cellular responses that overall are cell-type specific. Besides p53-dependent neuronal apoptosis, our findings that MEFs from compound Rad54/Parp-1 mutant mice display severe p53-mediated accelerated senescent phenotypes, and that features of senescence were detected in several neonatal tissues, strongly argue for senescence as the mechanism underlying synthetic lethal interactions of Rad54 and Parp-1 inactivation in vivo. Notably, many of the DNA repair pathways found deregulated in the cerebellum of Rad54-/-/Parp-1-/-/Ptc1+/- mutants have been reported to cause senescence; overexpression of Pttg1, the most upregulated gene in our settings, induces senescence in a p53-dependent fashion in normal fibroblasts [45], and chemical inhibition of DNA-PKcs leads to p53-dependent accelerated senescence after irradiation in vivo and in vitro [46].
In summary, our data contribute to the understanding of the interplay of Rad54 and Parp-1 in vivo by showing that their combined genetic disruption caused a marked growth delay culminating in a perinatal lethal phenotype. We identified accumulation of DNA damage and enhanced apoptotic cell death as a driving mechanism for growth retardation during development of the cerebellum in Rad54/Parp-1 compound mutants. Mechanistically, NHEJ was selectively repressed through downregulation of DNA-PKcs. Concomitant downregulation of Atm was also identified, directly involving transcriptional repression of PI3K signaling in synthetic lethality caused by Rad54 and Parp-1 deficiency. Finally, combined inactivation of Rad54 and Parp-1 caused accumulation of p53 and p21, which resulted in enhanced senescence in MEFs and several mouse tissues from Rad54-/-/Parp-1-/-/Ptc1+/- mutants. The p53-depedent apoptotic/senescent phenotypes is also potentially tumor protective; in fact, inactivation of one copy of Parp-1 in Rad54-/-/Ptc1+/- mutants suppressed radiation-induced MB tumorigenesis, identifying haplosufficient interactions also during neoplastic growth. Investigating the functional interplay between the HR factor Rad54 and Parp-1 in vivo during development and oncogenesis may help delineate the molecular mechanisms of lethal synthetic interactions and guide more successful clinical translation.
MATERIAL AND METHODS
Animal breeding
Mice lacking one Ptc1 allele (Ptc1neo6-7/+, named Ptc1+/- throughout the text) were generated through disruption of exons 6 and 7 in 129/Sv embryonic stem cells [18], and maintained on CD1 background. Genotyping for Ptc1 was performed as described [18]. Ptc1+/- mice were crossed with Rad54-/- or Parp-1-/- mice on C57Bl/6 background [47,48,49], to produce an F1. F1 mice of selected genotypes, i.e. Rad54+/-/Ptc1+/- x Parp-1+/-/Ptc1+/+ and Rad54+/-/Ptc1+/+ x Parp-1+/-/Ptc1+/- were crossed to produce an F2 population. Finally Rad54+/-/Parp-1+/-/Ptc1+/+/ and Rad54+/-/Parp-1+/-/Ptc1+/- mice were intercrossed. Genotyping of mice was performed as described [21,49].
Animal treatment and irradiation
Mice were housed under conventional conditions with food and water available ad libitum and a 12-h light cycle. For long-term study, mice were irradiated at postnatal day 1 (P1) with 1 Gy of X rays. Irradiation was performed using a Gilardoni CHF 320 G X-ray generator (Gilardoni S.p.A., Mandello del Lario, Italy) operated at 250 kVp, 5mA, Half-Value Layer = 1.6 mm Cu (additional filtration of 2.0 mm Al and 0.5 mm Cu) [50]. The size of the experimental groups was chosen to ensure statistical relevance. Randomized methods were applied and experiments were performed blinded. All experiments were conducted according to the directive 2010/63/EU of the European Parlament. Animal studies were approved and permission was issued by “Ministero della Salute” (Approval number is 986/2015-PR).
Histological analysis and tumor quantification
Mice were observed daily for their lifespan. Upon decline of health (i.e., severe weight loss, paralysis, ruffling of fur, or inactivity), they were killed and autopsied. Normally appearing and tumor bearing brains were fixed in 10% buffered formalin or snap frozen in liquid nitrogen. Autopsied mice were also checked for the presence of any other tumor, and all tumor masses were collected and processed for histological analysis using standard methods. MB incidence was expressed as the percentage of mice with tumors.
Tissue collection
Brains from Ptc1+/-, Parp-1-/-/Ptc1+/-, Rad54-/-/Ptc1+/-, and Rad54-/-/Parp-1-/-/Ptc1+/- were collected at P1 and P7. Samples were fixed in 10% buffered formalin. For mRNA profiling and western blot analysis P1 cerebella were snap frozen in liquid nitrogen and stored at -80°C.
Microsatellite analysis at the Ptc1 locus in MB
DNA from MBs and normal tissue was extracted using Wizard SV Genomic DNA Purification System (Promega Corporation, Madison, WI). Polymerase chain reaction amplifications were performed as described previously. About 10-14 microsatellites spanning the length of chromosome (chr) 13 were used to examine tumor DNA in comparison with the corresponding genomic DNA [51].
Morphometric analysis
Cerebella external granule layer (EGL) and internal granule layer (IGL) total cross sectional areas were measured in at least 3 samples. The imaging software NIS-Elements BR 4.00.05 (Nikon, Instruments Europe B.V., Italy) was used for morphometric analyses.
Immunohistochemical analysis
Cerebella and other tissues paraffin sections were used for immunohistochemical analysis as described previously [50]. Antibodies used were Ki-67 (ab15580, Abcam, Cambridge, UK), cleaved caspase-3 (9664, Cell Signaling Technology, Danvers, MA) and γ-H2AX (#2577, Cell Signaling Technology) and NeuN (MAB377, Millipore, Billerica, MA).
Ki-67 quantification: Three to 7 sections from EGL of Ptc1+/-, Rad54-/-/Ptc1+/-, Parp-1-/-/Ptc1+/- and Rad54-/-/Parp-1-/-/Ptc1+/- mice were immunostained for Ki67. Images were captured by HistoFAXS software (TissueGnostics GmbH, Vienna, Austria) at 20x magnification. Specific regions of interest (EGL) were analyzed with HistoQuest software (TissueGnostics) for automatic color separation and quantification. Expression levels were evaluated as stained area per µm2.
γ-H2AX and cleaved-caspase-3 quantification: Three to 5 sections from EGL of Ptc1+/-, Rad54-/-/Ptc1+/-, Parp-1-/-/Ptc1+/- and Rad54-/-/Parp-1-/-/Ptc1+/- mice were immunostained for γ-H2AX or cleaved caspase-3. expression levels were evaluated counting stained positive cells/ EGL area (mm2).
NeuN quantification: Three to 5 cerebellum sections of Ptc1+/-, Parp-1-/-/Ptc1+/-, Rad54-/-/Ptc1+/- and Rad54-/-/Parp-1-/-/Ptc1+/- mice were immunostained for NeuN. Expression levels of NeuN were evaluated as the percentage of stained positive cells in a fixed area (10000 µm2) of folia VIII for each cerebellum.
Preparation of mouse embryonic fibroblast (MEFs)
MEFs (two independent embryos per genotype) were generated from embryos at gestational day 13.5 (E13.5) according to standard protocols [52]. Briefly, the whole embryo was disaggregated using a mechanical disaggregator system (Medimax, CTSV, Bruino, TO, Italy) and grown in DMEM with 15% fetal bovine serum (FBS), 100 units/ml penicillin, 100 mg/ml streptomycin, and 2mM L-glutammine, in a humidified incubator with 5% CO2. All experiments were performed with cells that had been subjected to 2 passages.
Sulforhodamine B (SRB) assay
For the growth curve SRB assay was performed as described previously [53]. The plate was analysed at 510 nm optical density in the VICTOR X3 Multilabel Plate Reader (PerkinElmer, Waltham, MA). Doubling time (Dt) was calculated with the formula: Dt = t log2 / (log n1 – log n0), where n0 is the number of cell at time t0, n1 is the number of cells at time t1 and t is the time in exponential growth phase required by cells to increase their number from n0 to n1 (t = t1 – t0).
SA-β-Gal staining
Cells were cultured for 24 h, and stained using the Senescence β-Galactosidase Staining Kit (Cell Signalling Technology). SA-β-Gal staining of MEFs was performed following manufacturer’s instructions. The Percentage of cells expressing SA-β-Gal was quantified by inspecting at least 1000 cells in at least two different cell lines for each genotype.
Immunoblotting
Appropriate amounts of proteins (30 µg or 60 µg), extracted from a pool of 3 P1-cerebella and MEFs were loaded and separated by SDS-PAGE. Proteins were electro transferred to PVDF or nitrocellulose membranes (Trans-Blot Turbo Transfer Pack, BIO-RAD Laboratories, Hercules, CA) at the Trans-Blot Turbo Transfer System (BIO-RAD Laboratories). After blocking, membranes were incubated over night at +4°C with primary antibodies against DNA-PKcs and p53 (ab70250 and ab31333, Abcam, Cambridge, UK), ATM, p21 (sc-23921 and sc-471, Santa Cruz Biotechnology, Santa Cruz, CA), anti-phospho-p53(Ser18) (9284, Cell Signaling Technology, Inc., Danvers, MA), β-actin and HSP70 (A5316 and H5147, Sigma-Aldrich, St Louis, MO). Membranes were probed for 1h at RT with appropriated HRP-conjugated secondary antibodies (Santa Cruz Biotechnology). Immunoreactive bands were visualized using Amersham ECL Prime WB detection reagent (GE Healthcare Europe, Milan, Italy). Images were acquired using a Image 6 quant LAS 500 (GE Healthcare Europe), and densitometric analysis was performed using ImageJ software.
mRNA extraction and reverse transcription
Three to 7 cerebella were pooled for each genotype. mRNA was extracted with RNeasy Microarray Tissue kit (Qiagen, Germany) in accordance with manufacturer’s instructions. Reverse transcription was accomplished with the RT2 First Strand kit (Qiagen, Germany) as suggested by manufacturer.
RT2 Profiler PCR Array
RT2 RNA QC PCR Array (SaBiosciences, Qiagen, Germany) was used to test for RNA quality and inhibitors of RT-PCR analysis. For quantitative comparison of mRNA levels, real-time PCR was performed using RT2 Profiler PCR Arrays Mouse DNA Damage (SaBiosciences, Qiagen). For each genotype, two assays were carried out. Gene expression was related to the mean expression of all five housekeeping genes included in the array. Only Ct values <35 were included in the calculations. Data presented are averages of two independent experiments.
Prkdc and Atm genes were further validated by SYBR Green real-time PCR (SaBiosciences, Qiagen). The PCR primers for mouse Atm (PPM03454C-200 RT² qPCR Primer Assay ) and Prkdc (PPM03711E-200 RT² qPCR Primer Assay) were purchased from SABiosciences, Qiagen and real-time PCR were performed according to manufacturer’s instructions. Data represent the average of three independent experiments.
Bioinformatic analysis
Hierarchical clustering analysis was performed to evaluate similarities among the three genotypes when compared to Ptc1+/-mice. Euclidean distance matrices were performed in R (http://www.r-project.org/) using the fold change of each gene. Heat map visualization generated by R package “gplots” was used to show RT2 Profiler PCR Arrays Mouse DNA Damage. To estimate genotype-dependent pathway perturbations, the Signaling Protein Impact Analysis (SPIA) [54] algorithm based on KEGG (Kyoto Encyclopedia of Genes and Genomes) database was applied using the online tool Graphite (http://graphiteweb.bio.unipd.it/index.html). Significantly perturbed genes obtained from SPIA were analyzed by the Cytoscape tool [55] to evaluate the most perturbed pathways.
Statistical analysis
Appropriate statistical tests were used. Analyses were performed using GraphPad Prism version 4.02 for Windows (GraphPad Software, San Diego, CA). Statistical comparisons were made using Student’s t-test. Kaplan-Meier survival curves were compared and log rank test P values were calculated. P values ≤ 0.05 were considered statistically significant.
Conflicts of interest
The authors declare that they have no conflict of interest.
GRANT SUPPORT
Supported in part by Investigator Grant 15234 from Associazione Italiana Ricerca sul Cancro (AIRC). PG is a PhD student in “Molecular, Cellular and Environmental Biology”, accounting at Department of Sciences, Roma Tre University, Rome, Italy.
Editorial Note
This papers has been accepteds based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005; 434: 917-921.
2. Bryant HE, Petermann E, Schultz N, Jemth AS, Loseva O, Issaeva N, Johansson F, Fernandez S, McGlynn P, Helleday T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009; 28: 2601-2615.
3. Arnaudeau C, Lundin C, Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol. 2001; 307: 1235-1245.
4. Helleday T. Pathways for mitotic homologous recombination in mammalian cells. Mutat Res. 2003; 532: 103-115.
5. Ko HL, Ren EC. Functional Aspects of PARP1 in DNA Repair and Transcription. Biomolecules. 2012; 2: 524-548.
6. Jagtap P, Szabó C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005; 4: 421-440.
7. Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010; 10: 293-301.
8. Shall S, de Murcia G. Poly(ADP-ribose) polymerase-1: what have we learned from the deficient mouse model? Mutat Res. 2000; 460: 1-15.
9. Eisen JA, Sweder KS, Hanawalt PC. Evolution of the SNF2 family of proteins: subfamilies with distinct sequences and functions. Nucleic Acids Res. 1995; 23: 2715–2723.
10. Clever B, Interthal H, Schmuckli-Maurer J, King J, Sigrist M, Heyer WD. Recombinational repair in yeast: functional interactions between Rad51 and Rad54 proteins. Embo J. 1997; 16: 2535–2544.
11. Raschle M, Van Komen S, Chi P, Ellenberger T, Sung P. Multiple interactions with the Rad51 recombinase govern the homologous recombination function of Rad54. J Biol Chem. 2004; 279: 51973–51980.
12. Petukhova G, Stratton S, Sung P. Catalysis of homologous DNA pairing by yeast Rad51 and Rad54 proteins. Nature. 1998; 393: 91–94.
13. Bugreev DV, Mazina OM, Mazin AV. Rad54 protein promotes branch migration of Holliday junctions. Nature. 2006; 442: 590–593.
14. Hiramoto T, Nakanishi T, Sumiyoshi T, Fukuda T, Matsuura S, Tauchi H, Komatsu K, Shibasaki Y, Inui H, Watatani M, Yasutomi M, Sumii K, Kajiyama G, et al. Mutations of a novel human RAD54 homologue, RAD54B, in primary cancer. Oncogene. 1999; 18: 3422–3426.
15. Matsuda M, Miyagawa K, Takahashi M, Fukuda T, Kataoka T, Asahara T, Inui H, Watatani M, Yasutomi M, Kamada N, Dohi K, Kamiya K. Mutations of a novel human RAD54 homologue, RAD54B, in primary cancer. Oncogene. 1999; 18: 3427–3430.
16. Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, O’Connor KW, Konstantinopoulos PA, Elledge SJ, Boulton SJ, Yusufzai T, D’Andrea AD. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature. 2015; 518: 258-262.
17. Goodrich LV, Milenković L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997; 277: 1109-1113.
18. Hahn H, Wojnowski L, Zimmer AM, Hall J, Miller G, Zimmer A. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nat Med. 1998; 4: 619-622.
19. Pazzaglia S, Mancuso M, Atkinson MJ, Tanori M, Rebessi S, Majo VD, Covelli V, Hahn H, Saran A. High incidence of medulloblastoma following X-ray-irradiation of newborn Ptc1 heterozygous mice. Oncogene. 2002; 21: 7580-7584.
20. Pazzaglia S, Tanori M, Mancuso M, Rebessi S, Leonardi S, Di Majo V, Covelli V, Atkinson MJ, Hahn H, Saran A. Linking DNA damage to medulloblastoma tumorigenesis in patched heterozygous knockout mice. Oncogene. 2006; 25: 1165-1173.
21. Tanori M, Pasquali E, Leonardi S, Giardullo P, Di Majo V, Taccioli G, Essers J, Kanaar R, Mullenders LH, Atkinson MJ, Mancuso M, Saran A, Pazzaglia S. Opposite modifying effects of HR and NHEJ deficiency on cancer risk in Ptc1 heterozygous mouse cerebellum. Oncogene. 2011; 30: 4740-4749.
22. Tanori M, Mancuso M, Pasquali E, Leonardi S, Rebessi S, Di Majo V, Guilly MN, Giangaspero F, Covelli V, Pazzaglia S, Saran A. PARP-1 cooperates with Ptc1 to suppress medulloblastoma and basal cell carcinoma. Carcinogenesis. 2008; 29: 1911-1919.
23. Essers J, van Steeg H, de Wit J, Swagemakers SM, Vermeij M, Hoeijmakers JH, Kanaar R. Homologous and non-homologous recombination differentially affect DNA damage repair in mice. EMBO J. 2000; 19: 1703-1710.
24. Pazzaglia S, Tanori M, Mancuso M, Gessi M, Pasquali E, Leonardi S, Oliva MA, Rebessi S, Di Majo V, Covelli V, Giangaspero F, Saran A. Two-hit model for progression of medulloblastoma preneoplasia in Patched heterozygous mice. Oncogene. 2006; 25: 5575-5580.
25. Jones RM, Petermann E. Replication fork dynamics and the DNA damage response. Biochem J. 2012; 443: 13-26.
26. Petermann E, Helleday T. Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol. 2010; 11: 683-687.
27. Chao C, Saito S, Anderson CW, Appella E, Xu Y. Phosphorylation of murine p53 at ser-18 regulates the p53 responses to DNA damage. Proc Natl Acad Sci USA. 2000; 97: 11936-11941.
28. Nijman SM, Friend SH. Cancer. Potential of the synthetic lethality principle. Science. 2013; 342: 809-811.
29. McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O’Connor MJ, Tutt AN, Zdzienicka MZ, Smith GC, Ashworth A. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006; 66: 8109-8115.
30. McLornan DP, List A, Mufti GJ. Applying synthetic lethality for the selective targeting of cancer. N Engl J Med. 2014; 371: 1725-1735.
31. Anheim M. Autosomal recessive cerebellar ataxias. Rev Neurol (Paris). 2011; 167: 372-384.
32. Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci U S A. 2011; 108: 3406-3411.
33. Abraham RT. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair (Amst). 2004; 3: 883-887.
34. Shiloh Y.. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003, 3: 155-168.
35. Sekiguchi J, Ferguson DO, Chen HT, Yang EM, Earle J, Frank K, Whitlow S, Gu Y, Xu Y, Nussenzweig A, Alt FW. Genetic interactions between ATM and the nonhomologous end-joining factors in genomic stability and development. Proc Natl Acad Sci U S A. 2001; 98: 3243-3248.
36. Gurley KE, Kemp CJ. Synthetic lethality between mutation in Atm and DNA-PK(cs) during murine embryogenesis. Curr Biol. 2001; 11: 191-194.
37. Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmañà J, Rajendran A, Papa A, Spencer K, Lyssiotis CA, Nardella C, Pandolfi PP, Baselga J, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012; 2: 1048-1063.
38. Loughery J, Cox M, Smith LM, Meek DW. Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters. Nucleic Acids Res. 2014; 42: 7666-7680.
39. Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009; 137: 609-622.
40. Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, Seidl KJ, Sekiguchi JM, Rathbun GA, Swat W, Wang J, Bronson RT, Malynn BA, Bryans M, et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998; 95: 891-902.
41. Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C, Manis JP, Horner J, DePinho RA, Alt FW. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell. 2000; 5: 993-1002.
42. Lim DS, Hasty P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol Cell Biol. 1996; 16: 7133-7143.
43. Danilova N, Kumagai A, Lin J. p53 upregulation is a frequent response to deficiency of cell-essential genes. PLoS One. 2010; 5: e15938.
44. Lozano G. Mouse models of p53 functions. Cold Spring Harb Perspect Biol. 2010; 2: a001115.
45. Hsu YH, Liao LJ, Yu CH, Chiang CP, Jhan JR, Chang LC, Chen YJ, Lou PJ, Lin JJ. Overexpression of the pituitary tumor transforming gene induces p53-dependent senescence through activating DNA damage response pathway in normal human fibroblasts. J Biol Chem. 2010; 285: 22630-22638.
46. Azad A, Jackson S, Cullinane C, Natoli A, Neilsen PM, Callen DF, Maira SM, Hackl W, McArthur GA, Solomon B. Inhibition of DNA-dependent protein kinase induces accelerated senescence in irradiated human cancer cells. Mol Cancer Res. 2011; 9: 1696-1707.
47. Essers J, Hendriks RW, Swagemakers SM, Troelstra C, de Wit J, Bootsma D, Hoeijmakers JH, Kanaar R. Disruption of mouse RAD54 reduces ionizing radiation resistance and homologous recombination. Cell. 1997; 89: 195-204.
48. Wang ZQ, Auer B, Stingl L, Berghammer H, Haidacher D, Schweiger M, Wagner EF. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995; 9: 509-520.
49. Tong WM, Hande MP, Lansdorp PM, Wang ZQ. DNA strand break-sensing molecule poly(ADP-Ribose) polymerase cooperates with p53 in telomere function, chromosome stability, and tumor suppression. Mol Cell Biol. 2001; 21: 4046-4054.
50. Tanori M, Pasquali E, Leonardi S, Casciati A, Giardullo P, De Stefano I, Mancuso M, Saran A, Pazzaglia S. Developmental and oncogenic radiation effects on neural stem cells and their differentiating progeny in mouse cerebellum. Stem Cells. 2013; 31:2506-2516.
51. Pazzaglia S, Mancuso M, Tanori M, Atkinson MJ, Merola P, Rebessi S, Di Majo V, Covelli V, Hahn H, Saran A. Modulation of patched-associated susceptibility to radiation induced tumorigenesis by genetic background. Cancer Res. 2004; 64: 3798-806.
52. Espejel S, Franco S, Sgura A, Gae D, Bailey SM, Taccioli GE, Blasco MA. Functional interaction between DNA-PKcs and telomerase in telomere length maintenance. EMBO J. 2002; 21: 6275-6287.
53. Khongkow M, Olmos Y, Gong C, Gomes AR, Monteiro LJ, Yagüe E, Cavaco TB, Khongkow P, Man EP, Laohasinnarong S, Koo CY, Harada-Shoji N, Tsang JW, et al. SIRT6 modulates paclitaxel and epirubicinresistance and survival in breast cancer. Carcinogenesis. 2013; 34: 1476-1486.
54. Tarca AL, Bhatti G, Romero R. A comparison of gene set analysis methods in terms of sensitivity, prioritization and specificity. PLoS One. 2013; 8: e79217.
55. Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pagès F, Trajanoski Z, Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009; 25: 1091-1093.